Le langage universel du vivant c'est la chimie qui se déroule derrière.

Bernard OFFMANN
Professeur Université
section 64
| Équipe : |
Thèmes de recherche
Développements méthodologiques en bioinformatique structurale
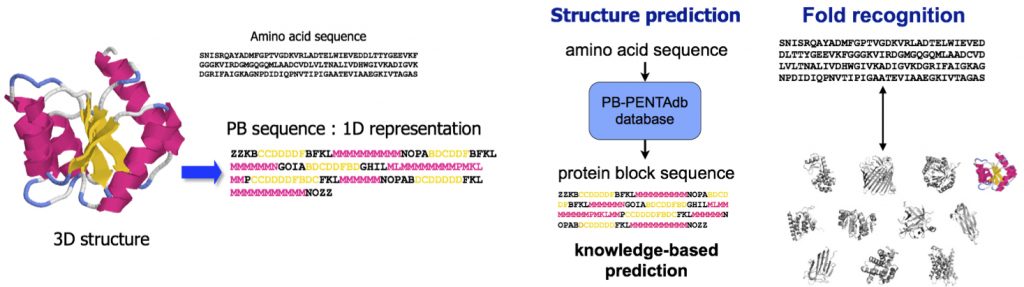
- approches autour d’un alphabet structural (les blocs protéiques) pour l’analyse et la prédiction des structures des protéines
- approches computationnelles pour l’évolution dirigée et l’ingénierie des protéines
- caractérisation structurale et fonctionnelle de grandes familles de protéines
Les applications
- développement de méthodes et logiciels pour la reconnaissance du repliement des protéines à l’échelle des génomes
- le protein design
- la caractérisation et l’ingénierie des enzymes actifs sur les sucres
- les bases structurales de l’olfaction dans le vivant notamment chez le moustique
Mes logiciels et bases de données
- iMutate : simulation de mutagénèse dirigée
- PB-kPRED : prédiction de squelette cartonnée des protéines à l’aide des blocs protéiques
- FORSA : reconnaissance du repliement des protéines à l’aide des blocs protéiques
- PENTAPEPT : base de données de pentapeptides constitutifs des structures des protéines
- PBE : Protein Block Expert (plateforme d’outils autour des blocs protéiques)
- PB-Frag : Fragments pour la modélisation ab initio des protéines
- mOBPdb : Mosquito odorant binding proteins database
Projets



Parcours universitaire
2006 – Habilitation à diriger des recherches. Université de La Réunion. Nouvelles approches en bioinformatique pour l’analyse et la prédiction des structures des protéines.
2000 – Doctorat en biochimie et biologie moléculaire. Université de La Réunion. Caractérisation et analyse génétique de la résistance de la canne à sucre à Xanthomonas albilineans
1997 – DEA en Analyse du Génome et Modélisation Moléculaire. Université Denis Diderot, Paris VII.
1996 – Maîtrise es Sciences et Techniques (MST), Chimie-Biologie : Valorisation chimique et biologique du végétal. Université de La Réunion.
1993 – DEUG SNV Biologie, Biochimie, Chimie. Université d’Avignon et des Pays du Vaucluse.
Publications
1 publication
Álvarez-Sánchez, Elena; Offmann, Bernard; Huet, Simon; Téletchéa, Stéphane
Energetics Decomposition of Sac7d:DNA Decrypts Amino Acids Role Without DNA Sequence Selectivity Article de journal
Dans: Journal Of Molecular Recognition, vol. 39, iss. 1, p. e70021, 2026.
@article{TeletcheaSac7d2025,
title = {Energetics Decomposition of Sac7d:DNA Decrypts Amino Acids Role Without DNA Sequence Selectivity},
author = {Elena Álvarez-Sánchez and Bernard Offmann and Simon Huet and Stéphane Téletchéa},
editor = {Wiley},
doi = {10.1002/jmr.70021},
year = {2026},
date = {2026-01-05},
urldate = {2025-12-15},
journal = {Journal Of Molecular Recognition},
volume = {39},
issue = {1},
pages = {e70021},
abstract = {Sac7d is a 7 kDa protein belonging to the class of the small chromosomal proteins from archeon Sulfolobus acidocaldarius. Sac7d was discovered in 1974 in Yellowstone National Parks geysers, and studied extensively since then for its remarkable stability at large pH and temperature ranges. Sac7d binds to the DNA minor groove, thereby protecting the host genome from extreme conditions by increasing the DNA melting temperature. In this study, we analyzed the Sac7d-DNA complex using 1 μs molecular dynamics simulations. The interaction energy of the interface was decomposed using Molecular Mechanics with Generalized Born Surface Area (MM/GBSA) to determine the residues that contributed most significantly to DNA binding. Out of 12 amino acids considered essential for DNA binding, three were newly identified in this study and had not been previously reported. One of these new amino acids, R63, may be involved in a dynamic protein-DNA interaction. The simulations performed also revealed a sliding motion of Sac7d over double-stranded DNA, suggesting a minimal sequence dependence interaction. Our analysis thus provides novel insights into how the Sac7d chaperones allow to protect DNA from degradation in extreme conditions. },
keywords = {},
pubstate = {published},
tppubtype = {article}
}
3 publications
Dhingra, Surbhi; Téletchéa, Stéphane; Sowdhamini, Ramanathan; Sanejouand, Yves-Henri; Brevern, Alexandre G.; Cadet, Frédéric; Offmann, Bernard
Using protein blocks to build custom fragment libraries from protein structures Article de journal À paraître
Dans: Biochimie, À paraître, ISSN: 0300-9084.
@article{DHINGRA2025,
title = {Using protein blocks to build custom fragment libraries from protein structures},
author = {Surbhi Dhingra and Stéphane Téletchéa and Ramanathan Sowdhamini and Yves-Henri Sanejouand and Alexandre G. Brevern and Frédéric Cadet and Bernard Offmann},
url = {https://www.sciencedirect.com/science/article/pii/S0300908425001907},
doi = {https://doi.org/10.1016/j.biochi.2025.08.011},
issn = {0300-9084},
year = {2025},
date = {2025-08-13},
urldate = {2025-01-01},
journal = {Biochimie},
abstract = {The remarkable structural diversity of modern proteins reflects millions of years of evolution, during which sequence space has expanded while many structural features remain conserved. This conservation is evident not only among homologous proteins but also in the recurrence of supersecondary motifs across unrelated proteins, underscoring the abundance and robustness of these structural units. Here, we present a novel pipeline for generating customized protein fragment libraries using protein blocks (PBs)—a structural alphabet that encodes local backbone conformations. Our method efficiently extracts structurally similar fragments from a curated, non-redundant protein structure database by converting three-dimensional structures into one-dimensional PB sequences. By integrating predicted PB sequences with the PB-ALIGN and PB-kPRED tools, our approach identifies relevant fragments independently of sequence homology. Fragment quality is further assessed using a new scoring function that combines secondary structure similarity and PB alignment metrics. The resulting libraries contain fragments of at least seven PBs (11 amino acid residues), covering over 70% of the local backbone structure. Our results demonstrate that PBs enable efficient mining of high-quality structural fragments from diverse protein spaces, including proteins with disordered regions. The pipeline is accessible as an online tool (PB-Frag, http://pbpred-us2b.univ-nantes.fr/pbfrag).},
keywords = {},
pubstate = {forthcoming},
tppubtype = {article}
}
Offmann, Bernard; Brevern, Alexandre G.
A 25-year journey with protein blocks: Unveiling the versatility of a structural alphabet Article de journal
Dans: Biochimie, 2025, ISSN: 0300-9084.
@article{OFFMANN2025,
title = {A 25-year journey with protein blocks: Unveiling the versatility of a structural alphabet},
author = {Bernard Offmann and Alexandre G. Brevern},
url = {https://www.sciencedirect.com/science/article/pii/S0300908425001750},
doi = {https://doi.org/10.1016/j.biochi.2025.08.007},
issn = {0300-9084},
year = {2025},
date = {2025-08-09},
urldate = {2025-01-01},
journal = {Biochimie},
abstract = {Protein Blocks (PBs) represent a widely used structural alphabet that enables the approximation and analysis of local protein conformations through 16 prototype fragments defined by dihedral angles. Initially developed to overcome the limitations of classical secondary structure definitions, PBs provide a powerful tool for understanding protein structure, dynamics, and function. Their applications span structural annotation, protein fold superimposition and recognition, sequence-based prediction and molecular dynamics analysis. Notably, PBs facilitate the distinction between rigid, flexible, and disordered regions via an entropy-based index (Neq), offering insights into protein flexibility and intrinsic disorder. Their integration with deep learning has dramatically improved predictive performance, and their utility has been demonstrated in diverse contexts such as integrin polymorphisms, VHH variability and AlphaFold structure analysis. As a robust and adaptable framework, PBs remain central in modern structural bioinformatics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fredslund, Folmer; Goux, Marine; Offmann, Bernard; Demonceaux, Marie; André-Miral, Corinne; Welner, Ditte; Teze, David
Crystal structure of the sucrose phosphorylase from Alteromonas mediterranea shows a loop transition in the active site Article de journal
Dans: Acta Crystallographica Section F, vol. 81, no. 7, p. 306–310, 2025.
@article{Fredslund:us5158,
title = {Crystal structure of the sucrose phosphorylase from Alteromonas mediterranea shows a loop transition in the active site},
author = {Folmer Fredslund and Marine Goux and Bernard Offmann and Marie Demonceaux and Corinne André-Miral and Ditte Welner and David Teze},
url = {https://doi.org/10.1107/S2053230X25004327},
doi = {10.1107/S2053230X25004327},
year = {2025},
date = {2025-07-01},
urldate = {2025-07-01},
journal = {Acta Crystallographica Section F},
volume = {81},
number = {7},
pages = {306–310},
abstract = {Sucrose phosphorylases are essential enzymes regulating sucrose metabolism, and it has been shown that a loop rearrangement is essential to their catalytic cycle. Crystal structures of only six sucrose phosphorylase enzymes are available. Here, we present the crystal structure of a sucrose phosphorylase from a proteobacterium, ıt Alteromonas mediterranea, at 2.15Å resolution. The available sucrose phosphorylase structures have shown that an important conformational change occurs during the catalytic cycle or upon mutagenesis. Interestingly, our data present clear indications of the two major conformations in the same crystal.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2 publications
Goux, Marine; Demonceaux, Marie; Hendrickx, Johann; Solleux, Claude; Lormeau, Emilie; Fredslund, Folmer; Tezé, David; Offmann, Bernard; André-Miral, Corinne
Sucrose phosphorylase from Alteromonas mediterranea: structural insight into the regioselective α-glucosylation of (+)-catechin Article de journal
Dans: Biochimie, 2024.
@article{Goux2023.04.11.536264,
title = {Sucrose phosphorylase from Alteromonas mediterranea: structural insight into the regioselective α-glucosylation of (+)-catechin},
author = {Marine Goux and Marie Demonceaux and Johann Hendrickx and Claude Solleux and Emilie Lormeau and Folmer Fredslund and David Tezé and Bernard Offmann and Corinne André-Miral},
url = {https://www.biorxiv.org/content/10.1101/2023.04.11.536264v2
hal-04095395v2 },
doi = {10.1016/j.biochi.2024.01.004},
year = {2024},
date = {2024-01-09},
urldate = {2024-01-09},
journal = {Biochimie},
publisher = {Cold Spring Harbor Laboratory},
abstract = {Sucrose phosphorylases, through transglycosylation reactions, are interesting enzymes that can transfer regioselectively glucose from sucrose, the donor substrate, onto acceptors like flavonoids to form glycoconjugates and hence modulate their solubility and bioactivity. Here, we report for the first time the structure of sucrose phosphorylase from the marine bacteria Alteromonas mediterranea (AmSP) and its enzymatic properties. Kinetics of sucrose hydrolysis and transglucosylation capacities on (+)-catechin were investigated. Wild-type enzyme (AmSP-WT) displayed high hydrolytic activity on sucrose and was devoid of transglucosylation activity on (+)-catechin. Two variants, AmSP-Q353F and AmSP-P140D catalysed the regiospecific transglucosylation of (+)-catechin: 89 % of a novel compound (+)-catechin-4′-O-α-d-glucopyranoside (CAT-4′) for AmSP-P140D and 92 % of (+)-catechin-3′-O-α-d-glucopyranoside (CAT-3′) for AmSP-Q353F. The compound CAT-4′ was fully characterized by NMR and mass spectrometry. An explanation for this difference in regiospecificity was provided at atomic level by molecular docking simulations: AmSP-P140D was found to preferentially bind (+)-catechin in a mode that favours glucosylation on its hydroxyl group in position 4′ while the binding mode in AmSP-Q353F favoured glucosylation on its hydroxyl group in position 3’.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Graton, Jérôme; Goupille, Anaïs; Ferré, Tanguy; Offmann, Bernard; André-Miral, Corinne; Questel, Jean-Yves Le
Antioxidant properties of catechin and its 3′O-α-glucoside: Insights from computational chemistry calculations Article de journal
Dans: Computational and Theoretical Chemistry, vol. 1236, p. 114608, 2024, ISSN: 2210-271X.
@article{GRATON2024114608,
title = {Antioxidant properties of catechin and its 3′O-α-glucoside: Insights from computational chemistry calculations},
author = {Jérôme Graton and Anaïs Goupille and Tanguy Ferré and Bernard Offmann and Corinne André-Miral and Jean-Yves Le Questel},
url = {https://www.sciencedirect.com/science/article/pii/S2210271X24001476
https://hal.science/hal-04610796v1},
doi = {https://doi.org/10.1016/j.comptc.2024.114608},
issn = {2210-271X},
year = {2024},
date = {2024-01-01},
urldate = {2024-01-01},
journal = {Computational and Theoretical Chemistry},
volume = {1236},
pages = {114608},
abstract = {Density functional theory (DFT) calculations were used to investigate the conformational landscape of catechin and one of its main glucoside derivative (catechin-3′ O- α −glucopyranoside), and to determine the corresponding antioxidant properties. These investigations were carried out in benzene and water using the SMD universal continuum solvation model. Both properties were found to be significantly affected. The structures are characterized in both solvents by strong intramolecular hydrogen bonds (IMHB). In an apolar environment, Hydrogen Atom Transfer (HAT) is by far favored whereas in water the Sequential Proton Loss Electron Transfer (SPLET) mechanism is strongly preferred. In benzene, the catechin fragment has the best antioxidant character (from 27 kJ/mole) whereas in polar surroundings, the glucoside derivative has a slightly better antiradical activity (from 5 kJ/mole). Our results confirm the key role of the 3′-OH and 4′-OH groups of the catechole ring in these properties.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
3 publications
Demonceaux, Marie; Goux, Marine; Schimith, Lucia Emanueli; Santos, Michele Goulart Dos; Hendrickx, Johann; Offmann, Bernard; André-Miral, Corinne
Enzymatic synthesis, characterization and molecular docking of a new functionalized polyphenol: Resveratrol-3, 4’-⍺-diglucoside Article de journal
Dans: Results in Chemistry, p. 100956, 2023.
@article{demonceaux2023enzymatic,
title = {Enzymatic synthesis, characterization and molecular docking of a new functionalized polyphenol: Resveratrol-3, 4’-⍺-diglucoside},
author = {Marie Demonceaux and Marine Goux and Lucia Emanueli Schimith and Michele Goulart Dos Santos and Johann Hendrickx and Bernard Offmann and Corinne André-Miral},
url = {https://www.sciencedirect.com/science/article/pii/S2211715623001959},
doi = {10.1016/j.rechem.2023.100956},
year = {2023},
date = {2023-05-16},
urldate = {2023-05-16},
journal = {Results in Chemistry},
pages = {100956},
publisher = {Elsevier},
abstract = {Transglucosylation of resveratrol by the Q345F variant of sucrose phosphorylase from Bifidobacterium adolescentis (BaSP) was extensively studied during the last decade. Indeed, Q345F is able to catalyze the synthesis of resveratrol-3-O-⍺-D-glucoside (RES-3) with yield up to 97% using a cost-effective glucosyl donor, sucrose (Kraus et al., Chemical Communications, 53(90), 12182–12184 (2017)). Despite the fact that two further products were detectable in low amounts after glucoside synthesis, they were never identified. Here, we isolated and fully characterized one of those two minor products: resveratrol-3,4′-O-⍺-D-diglucoside (RES-3,4′). This original compound had never been described before. Using bioinformatics models, we successfully explained the formation of this diglucosylated product. Indeed, with RES-3 as acceptor substrate, Q345F is able to transfer a glucosyl moiety in position 4′-OH, what had been reported as impossible in the literature. The low yield observed is due to the steric hindrance into the catalytic site between RES-3 and residues Tyr132 and Tyr344. Nevertheless, the substrate orientation in the active site is favored by stabilizing interactions. Ring A of RES-3 bearing the diol moiety is stabilized by hydrogen bonds with residues Asp50, Arg135, Asn347 and Arg399. Hydroxyl group OH-4′ shares hydrogen bonds with the catalytic residues Asp192 and Glu232. Multiple hydrophobic contacts complete the stabilization of the substrate to favor the glucosylation at position 4′. Understanding of the mechanisms allowing the glucosylation at position 4′ of resveratrol will help the development of enzymatic tools to target and control the enzymatic synthesis of original ⍺-glucosylated polyphenols with high added value and better biodisponibility.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Demonceaux, Marie; Goux, Marine; Hendrickx, Johann; Solleux, Claude; Cadet, Frédéric; Lormeau, Émilie; Offmann, Bernard; André-Miral, Corinne
Regioselective glucosylation of (+)-catechin using a new variant of sucrose phosphorylase from Bifidobacterium adolescentis Article de journal
Dans: Organic & Biomolecular Chemistry, vol. 21, no. 11, p. 2307–2311, 2023.
@article{demonceaux2023regioselective,
title = {Regioselective glucosylation of (+)-catechin using a new variant of sucrose phosphorylase from Bifidobacterium adolescentis},
author = {Marie Demonceaux and Marine Goux and Johann Hendrickx and Claude Solleux and Frédéric Cadet and Émilie Lormeau and Bernard Offmann and Corinne André-Miral},
doi = {10.1039/D3OB00191A},
year = {2023},
date = {2023-02-22},
urldate = {2023-02-22},
journal = {Organic & Biomolecular Chemistry},
volume = {21},
number = {11},
pages = {2307--2311},
publisher = {Royal Society of Chemistry},
abstract = {Mutation Q345F in sucrose phosphorylase from Bifidobacterium adolescentis (BaSP) has shown to allow efficient (+)-catechin glucosylation yielding a regioisomeric mixture: (+)-catechin-3′-O-α-D-glucopyranoside, (+)-catechin-5-O-α-D-glucopyranoside and (+)-catechin-3′,5-O-α-D-diglucopyranoside with a ratio of 51 : 25 : 24. Here, we efficiently increased the control of (+)-catechin glucosylation regioselectivity with a new variant Q345F/P134D. The same products were obtained with a ratio of 82 : 9 : 9. Thanks to bioinformatics models, we successfully explained the glucosylation favoured at the OH-3′ position due to the mutation P134D.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Mam, Bhavika; Tsitsanou, Katerina E.; Liggri, Panagiota G. V.; Saitta, Francesca; Stamati, Evgenia C. V.; Mahita, Jarjapu; Leonis, Georgios; Drakou, Christina E.; Papadopoulos, Manthos; Arnaud, Philippe; Offmann, Bernard; Fessas, Dimitrios; Sowdhamini, Ramanathan; Zographos, Spyros E.
Influence of pH on indole-dependent heterodimeric interactions between Anopheles gambiae odorant-binding proteins OBP1 and OBP4 Article de journal
Dans: International Journal of Biological Macromolecules, vol. 245, p. 125422, 2023, ISSN: 0141-8130.
@article{MAM2023125422,
title = {Influence of pH on indole-dependent heterodimeric interactions between Anopheles gambiae odorant-binding proteins OBP1 and OBP4},
author = {Bhavika Mam and Katerina E. Tsitsanou and Panagiota G. V. Liggri and Francesca Saitta and Evgenia C. V. Stamati and Jarjapu Mahita and Georgios Leonis and Christina E. Drakou and Manthos Papadopoulos and Philippe Arnaud and Bernard Offmann and Dimitrios Fessas and Ramanathan Sowdhamini and Spyros E. Zographos},
url = {https://www.sciencedirect.com/science/article/pii/S0141813023023164},
doi = {https://doi.org/10.1016/j.ijbiomac.2023.125422},
issn = {0141-8130},
year = {2023},
date = {2023-01-01},
urldate = {2023-01-01},
journal = {International Journal of Biological Macromolecules},
volume = {245},
pages = {125422},
abstract = {Insect Odorant Binding Proteins (OBPs) constitute important components of their olfactory apparatus, as they are essential for odor recognition. OBPs undergo conformational changes upon pH change, altering their interactions with odorants. Moreover, they can form heterodimers with novel binding characteristics. Anopheles gambiae OBP1 and OBP4 were found capable of forming heterodimers possibly involved in the specific perception of the attractant indole. In order to understand how these OBPs interact in the presence of indole and to investigate the likelihood of a pH-dependent heterodimerization mechanism, the crystal structures of OBP4 at pH 4.6 and 8.5 were determined. Structural comparison to each other and with the OBP4-indole complex (3Q8I, pH 6.85) revealed a flexible N-terminus and conformational changes in the α4-loop-α5 region at acidic pH. Fluorescence competition assays showed a weak binding of indole to OBP4 that becomes further impaired at acidic pH. Additional Molecular Dynamic and Differential Scanning Calorimetry studies displayed that the influence of pH on OBP4 stability is significant compared to the modest effect of indole. Furthermore, OBP1-OBP4 heterodimeric models were generated at pH 4.5, 6.5, and 8.5, and compared concerning their interface energy and cross-correlated motions in the absence and presence of indole. The results indicate that the increase in pH may induce the stabilization of OBP4 by increasing its helicity, thereby enabling indole binding at neutral pH that further stabilizes the protein and possibly promotes the creation of a binding site for OBP1. A decrease in interface stability and loss of correlated motions upon transition to acidic pH may provoke the heterodimeric dissociation allowing indole release. Finally, we propose a potential OBP1-OBP4 heterodimer formation/disruption mechanism induced by pH change and indole binding.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2 publications
Guo, Xia; Xuan, Ning; Liu, Guoxia; Xie, Hongyan; Lou, Qinian; Arnaud, Philippe; Offmann, Bernard; Picimbon, Jean-François
An Expanded Survey of the Moth PBP/GOBP Clade in Bombyx mori: New Insight into Expression and Functional Roles Article de journal
Dans: Frontiers in Physiology, vol. 12, p. 1701, 2021, ISSN: 1664-042X.
@article{10.3389/fphys.2021.712593,
title = {An Expanded Survey of the Moth PBP/GOBP Clade in Bombyx mori: New Insight into Expression and Functional Roles},
author = {Xia Guo and Ning Xuan and Guoxia Liu and Hongyan Xie and Qinian Lou and Philippe Arnaud and Bernard Offmann and Jean-François Picimbon},
url = {https://www.frontiersin.org/article/10.3389/fphys.2021.712593},
doi = {10.3389/fphys.2021.712593},
issn = {1664-042X},
year = {2021},
date = {2021-10-28},
urldate = {2021-01-01},
journal = {Frontiers in Physiology},
volume = {12},
pages = {1701},
abstract = {We studied the expression profile and ontogeny (from the egg stage through the larval stages and pupal stages, to the elderly adult age) of four OBPs from the silkworm moth Bombyx mori. We first showed that male responsiveness to female sex pheromone in the silkworm moth B. mori does not depend on age variation; whereas the expression of BmorPBP1, BmorPBP2, BmorGOBP1, and BmorGOBP2 varies with age. The expression profile analysis revealed that the studied OBPs are expressed in non-olfactory tissues at different developmental stages. In addition, we tested the effect of insecticide exposure on the expression of the four OBPs studied. Exposure to a toxic macrolide insecticide endectocide molecule (abamectin) led to the modulated expression of all four genes in different tissues. The higher expression of OBPs was detected in metabolic tissues, such as the thorax, gut, and fat body. All these data strongly suggest some alternative functions for these proteins other than olfaction. Finally, we carried out ligand docking studies and reported that PBP1 and GOBP2 have the capacity of binding vitamin K1 and multiple different vitamins.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gheyouche, Ennys; Bagueneau, Matthias; Loirand, Gervaise; Offmann, Bernard; Téletchéa, Stéphane
Structural Design and Analysis of the RHOA-ARHGEF1 Binding Mode: Challenges and Applications for Protein-Protein Interface Prediction Article de journal
Dans: Frontiers in Molecular Biosciences, vol. 8, p. 643728, 2021, ISSN: 2296-889X.
@article{gheyouche_structural_2021,
title = {Structural Design and Analysis of the RHOA-ARHGEF1 Binding Mode: Challenges and Applications for Protein-Protein Interface Prediction},
author = {Ennys Gheyouche and Matthias Bagueneau and Gervaise Loirand and Bernard Offmann and Stéphane Téletchéa},
doi = {10.3389/fmolb.2021.643728},
issn = {2296-889X},
year = {2021},
date = {2021-01-01},
journal = {Frontiers in Molecular Biosciences},
volume = {8},
pages = {643728},
abstract = {The interaction between two proteins may involve local movements, such as small side-chains re-positioning or more global allosteric movements, such as domain rearrangement. We studied how one can build a precise and detailed protein-protein interface using existing protein-protein docking methods, and how it can be possible to enhance the initial structures using molecular dynamics simulations and data-driven human inspection. We present how this strategy was applied to the modeling of RHOA-ARHGEF1 interaction using similar complexes of RHOA bound to other members of the Rho guanine nucleotide exchange factor family for comparative assessment. In parallel, a more crude approach based on structural superimposition and molecular replacement was also assessed. Both models were then successfully refined using molecular dynamics simulations leading to protein structures where the major data from scientific literature could be recovered. We expect that the detailed strategy used in this work will prove useful for other protein-protein interface design. The RHOA-ARHGEF1 interface modeled here will be extremely useful for the design of inhibitors targeting this protein-protein interaction (PPI).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
5 publications
Dhingra, Surbhi; Sowdhamini, Ramanathan; Sanejouand, Yves-Henri; Cadet, Frédéric; Offmann, Bernard
Customised fragment libraries for ab initio protein structure prediction using a structural alphabet Article de journal
Dans: arXiv:2005.01696, 2020.
@article{Dhingra2020,
title = {Customised fragment libraries for ab initio protein structure prediction using a structural alphabet},
author = {Surbhi Dhingra and Ramanathan Sowdhamini and Yves-Henri Sanejouand and Frédéric Cadet and Bernard Offmann},
url = {https://arxiv.org/pdf/2005.01696.pdf},
year = {2020},
date = {2020-05-01},
journal = {arXiv:2005.01696},
abstract = {Motivation: Computational protein structure prediction has taken over the structural community in past few decades, mostly focusing on the development of Template-Free modelling (TFM) or ab initio modelling protocols. Fragment-based assembly (FBA), falls under this category and is by far the most popular approach to solve the spatial arrangements of proteins. FBA approaches usually rely on sequence based profile comparison to generate fragments from a representative structural database. Here we report the use of Protein Blocks (PBs), a structural alphabet (SA) to perform such sequence comparison and to build customised fragment libraries for TFM. Results: We demonstrate that predicted PB sequences for a query protein can be used to search for high quality fragments that overall cover above 90% of the query. The fragments generated are of minimum length of 11 residues, and fragments that cover more than 30% of the query length were often obtained. Our work shows that PBs can serve as a good way to extract structurally similar fragments from a database of representatives of non-homologous structures and of the proteins that contain less ordered regions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ostafe, Raluca; Fontaine, Nicolas; Frank, David; Chong, Matthieu Ng Fuk; Prodanovic, Radivoje; Pandjaitan, Rudy; Offmann, Bernard; Cadet, Frédéric; Fischer, Rainer
One-shot optimization of multiple enzyme parameters: Tailoring glucose oxidase for pH and electron mediators Article de journal
Dans: Biotechnology and Bioengineering, vol. 117, no. 1, p. 17–29, 2020, ISSN: 10970290.
@article{Ostafe2020,
title = {One-shot optimization of multiple enzyme parameters: Tailoring glucose oxidase for pH and electron mediators},
author = {Raluca Ostafe and Nicolas Fontaine and David Frank and Matthieu {Ng Fuk Chong} and Radivoje Prodanovic and Rudy Pandjaitan and Bernard Offmann and Frédéric Cadet and Rainer Fischer},
doi = {10.1002/bit.27169},
issn = {10970290},
year = {2020},
date = {2020-01-01},
journal = {Biotechnology and Bioengineering},
volume = {117},
number = {1},
pages = {17--29},
abstract = {Enzymes are biological catalysts with many industrial applications, but natural enzymes are usually unsuitable for industrial processes because they are not optimized for the process conditions. The properties of enzymes can be improved by directed evolution, which involves multiple rounds of mutagenesis and screening. By using mathematical models to predict the structure–activity relationship of an enzyme, and by defining the optimal combination of mutations in silico, we can significantly reduce the number of bench experiments needed, and hence the time and investment required to develop an optimized product. Here, we applied our innovative sequence–activity relationship methodology (innov'SAR) to improve glucose oxidase activity in the presence of different mediators across a range of pH values. Using this machine learning approach, a predictive model was developed and the optimal combination of mutations was determined, leading to a glucose oxidase mutant (P1) with greater specificity for the mediators ferrocene–methanol (12-fold) and nitrosoaniline (8-fold), compared to the wild-type enzyme, and better performance in three pH-adjusted buffers. The kcat/KM ratio of P1 increased by up to 121 folds compared to the wild type enzyme at pH 5.5 in the presence of ferrocene methanol.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nagaraja, Anamya Ajjolli; Charton, Philippe; Cadet, Xavier F; Fontaine, Nicolas; Delsaut, Mathieu; Wiltschi, Birgit; Voit, Alena; Offmann, Bernard; Damour, Cedric; Grondin-Perez, Brigitte; Cadet, Frederic
A Machine Learning Approach for Efficient Selection of Enzyme Concentrations and Its Application for Flux Optimization Article de journal
Dans: Catalysts, vol. 10, no. 3, 2020, ISSN: 2073-4344.
@article{catal10030291,
title = {A Machine Learning Approach for Efficient Selection of Enzyme Concentrations and Its Application for Flux Optimization},
author = {Anamya Ajjolli Nagaraja and Philippe Charton and Xavier F Cadet and Nicolas Fontaine and Mathieu Delsaut and Birgit Wiltschi and Alena Voit and Bernard Offmann and Cedric Damour and Brigitte Grondin-Perez and Frederic Cadet},
url = {https://www.mdpi.com/2073-4344/10/3/291},
doi = {10.3390/catal10030291},
issn = {2073-4344},
year = {2020},
date = {2020-01-01},
journal = {Catalysts},
volume = {10},
number = {3},
abstract = {The metabolic engineering of pathways has been used extensively to produce molecules of interest on an industrial scale. Methods like gene regulation or substrate channeling helped to improve the desired product yield. Cell-free systems are used to overcome the weaknesses of engineered strains. One of the challenges in a cell-free system is selecting the optimized enzyme concentration for optimal yield. Here, a machine learning approach is used to select the enzyme concentration for the upper part of glycolysis. The artificial neural network approach (ANN) is known to be inefficient in extrapolating predictions outside the box: high predicted values will bump into a sort of “glass ceiling”. In order to explore this “glass ceiling” space, we developed a new methodology named glass ceiling ANN (GC-ANN). Principal component analysis (PCA) and data classification methods are used to derive a rule for a high flux, and ANN to predict the flux through the pathway using the input data of 121 balances of four enzymes in the upper part of glycolysis. The outcomes of this study are i. in silico selection of optimum enzyme concentrations for a maximum flux through the pathway and ii. experimental in vitro validation of the “out-of-the-box” fluxes predicted using this new approach. Surprisingly, flux improvements of up to 63% were obtained. Gratifyingly, these improvements are coupled with a cost decrease of up to 25% for the assay.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Liu, Guoxia; Xuan, Ning; Rajashekar, Balaji; Arnaud, Philippe; Offmann, Bernard; Picimbon, Jean-François
Comprehensive History of CSP Genes: Evolution, Phylogenetic Distribution and Functions Article de journal
Dans: Genes, vol. 11, no. 4, p. 413, 2020.
@article{liu2020comprehensive,
title = {Comprehensive History of CSP Genes: Evolution, Phylogenetic Distribution and Functions},
author = {Guoxia Liu and Ning Xuan and Balaji Rajashekar and Philippe Arnaud and Bernard Offmann and Jean-François Picimbon},
doi = {10.3390/genes11040413},
year = {2020},
date = {2020-01-01},
journal = {Genes},
volume = {11},
number = {4},
pages = {413},
publisher = {Multidisciplinary Digital Publishing Institute},
abstract = {In this review we present the developmental, histological, evolutionary and functional properties of insect chemosensory proteins (CSPs) in insect species. CSPs are small globular proteins folded like a prism and notoriously known for their complex and arguably obscure function(s), particularly in pheromone olfaction. Here, we focus on direct functional consequences on protein function depending on duplication, expression and RNA editing. The result of our analysis is important for understanding the significance of RNA-editing on functionality of CSP genes, particularly in the brain tissue.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Dhingra, Surbhi; Sowdhamini, Ramanathan; Cadet, Frédéric; Offmann, Bernard
A glance into the evolution of template-free protein structure prediction methodologies Article de journal
Dans: Biochimie, vol. 175, p. 85 - 92, 2020, ISSN: 0300-9084.
@article{DHINGRA202085,
title = {A glance into the evolution of template-free protein structure prediction methodologies},
author = {Surbhi Dhingra and Ramanathan Sowdhamini and Frédéric Cadet and Bernard Offmann},
url = {http://www.sciencedirect.com/science/article/pii/S0300908420300961},
doi = {https://doi.org/10.1016/j.biochi.2020.04.026},
issn = {0300-9084},
year = {2020},
date = {2020-01-01},
journal = {Biochimie},
volume = {175},
pages = {85 - 92},
abstract = {Prediction of protein structures using computational approaches has been explored for over two decades, paving a way for more focused research and development of algorithms in comparative modelling, ab intio modelling and structure refinement protocols. A tremendous success has been witnessed in template-based modelling protocols, whereas strategies that involve template-free modelling still lag behind, specifically for larger proteins (>150 a.a.). Various improvements have been observed in ab initio protein structure prediction methodologies overtime, with recent ones attributed to the usage of deep learning approaches to construct protein backbone structure from its amino acid sequence. This review highlights the major strategies undertaken for template-free modelling of protein structures while discussing few tools developed under each strategy. It will also briefly comment on the progress observed in the field of ab initio modelling of proteins over the course of time as seen through the evolution of CASP platform.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
6 publications
Nagaraja, Anamya Ajjolli; Fontaine, Nicolas; Delsaut, Mathieu; Charton, Philippe; Damour, Cedric; Offmann, Bernard; Grondin-Perez, Brigitte; Cadet, Frédéric
Flux prediction using artificial neural network (ANN) for the upper part of glycolysis Article de journal
Dans: PLOS ONE, vol. 14, no. 5, p. e0216178, 2019, ISSN: 1932-6203.
@article{AjjolliNagaraja2019,
title = {Flux prediction using artificial neural network (ANN) for the upper part of glycolysis},
author = {Anamya {Ajjolli Nagaraja} and Nicolas Fontaine and Mathieu Delsaut and Philippe Charton and Cedric Damour and Bernard Offmann and Brigitte Grondin-Perez and Frédéric Cadet},
editor = {Marie-Joelle Virolle},
url = {https://dx.plos.org/10.1371/journal.pone.0216178},
doi = {10.1371/journal.pone.0216178},
issn = {1932-6203},
year = {2019},
date = {2019-05-01},
journal = {PLOS ONE},
volume = {14},
number = {5},
pages = {e0216178},
publisher = {Public Library of Science},
abstract = {The selection of optimal enzyme concentration in multienzyme cascade reactions for the highest product yield in practice is very expensive and time-consuming process. The modelling of biological pathways is a difficult process because of the complexity of the system. The mathematical modelling of the system using an analytical approach depends on the many parameters of enzymes which rely on tedious and expensive experiments. The artificial neural network (ANN) method has been successively applied in different fields of science to perform complex functions. In this study, ANN models were trained to predict the flux for the upper part of glycolysis as inferred by NADH consumption, using four enzyme concentrations i.e., phosphoglucoisomerase, phosphofructokinase, fructose-bisphosphate-aldolase, triose-phosphate-isomerase. Out of three ANN algorithms, the neuralnet package with two activation functions, “logistic” and “tanh” were implemented. The prediction of the flux was very efficient: RMSE and R2 were 0.847, 0.93 and 0.804, 0.94 respectively for logistic and tanh functions using a cross validation procedure. This study showed that a systemic approach such as ANN could be used for accurate prediction of the flux through the metabolic pathway. This could help to save a lot of time and costs, particularly from an industrial perspective. The R-code is available at: https://github.com/DSIMB/ANN-Glycolysis-Flux-Prediction.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Ghosh, Pritha; Joshi, Adwait; Guita, Niang; Offmann, Bernard; Sowdhamini, Ramanathan
EcRBPome: A comprehensive database of all known E. coli RNA-binding proteins Article de journal
Dans: BMC Genomics, vol. 20, no. 1, p. 1–6, 2019, ISSN: 14712164.
@article{Ghosh2019,
title = {EcRBPome: A comprehensive database of all known E. coli RNA-binding proteins},
author = {Pritha Ghosh and Adwait Joshi and Niang Guita and Bernard Offmann and Ramanathan Sowdhamini},
doi = {10.1186/s12864-019-5755-5},
issn = {14712164},
year = {2019},
date = {2019-01-01},
journal = {BMC Genomics},
volume = {20},
number = {1},
pages = {1--6},
publisher = {BMC Genomics},
abstract = {The repertoire of RNA-binding proteins (RBPs) in bacteria play a crucial role in their survival, and interactions with the host machinery, but there is little information, record or characterisation in bacterial genomes. As a first step towards this, we have chosen the bacterial model system Escherichia coli, and organised all RBPs in this organism into a comprehensive database named EcRBPome. It contains RBPs recorded from 614 complete E. coli proteomes available in the RefSeq database (as of October 2018). The database provides various features related to the E. coli RBPs, like their domain architectures, PDB structures, GO and EC annotations etc. It provides the assembly, bioproject and biosample details of each strain, as well as cross-strain comparison of occurrences of various RNA-binding domains (RBDs). The percentage of RBPs, the abundance of the various RBDs harboured by each strain have been graphically represented in this database and available alongside other files for user download. To the best of our knowledge, this is the first database of its kind and we hope that it will be of great use to the biological community.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Chaaya, Nancy; Shahsavarian, Melody A; Maffucci, Irene; Friboulet, Alain; Offmann, Bernard; Léger, Jean Benoist; Rousseau, Sylvain; Avalle, Bérangère; Padiolleau-Lefèvre, Séverine
Genetic background and immunological status influence B cell repertoire diversity in mice Article de journal
Dans: Scientific Reports, vol. 9, no. 1, p. 1–7, 2019, ISSN: 20452322.
@article{Chaaya2019,
title = {Genetic background and immunological status influence B cell repertoire diversity in mice},
author = {Nancy Chaaya and Melody A Shahsavarian and Irene Maffucci and Alain Friboulet and Bernard Offmann and Jean Benoist Léger and Sylvain Rousseau and Bérang{è}re Avalle and Séverine Padiolleau-Lef{è}vre},
doi = {10.1038/s41598-019-50714-y},
issn = {20452322},
year = {2019},
date = {2019-01-01},
journal = {Scientific Reports},
volume = {9},
number = {1},
pages = {1--7},
abstract = {The relationship between the immune repertoire and the physiopathological status of individuals is essential to apprehend the genesis and the evolution of numerous pathologies. Nevertheless, the methodological approaches to understand these complex interactions are challenging. We performed a study evaluating the diversity harbored by different immune repertoires as a function of their physiopathological status. In this study, we base our analysis on a murine scFv library previously described and representing four different immune repertoires: i) healthy and naïve, ii) healthy and immunized, iii) autoimmune prone and naïve, and iv) autoimmune prone and immunized. This library, 2.6 × 109 in size, is submitted to high throughput sequencing (Next Generation Sequencing, NGS) in order to analyze the gene subgroups encoding for immunoglobulins. A comparative study of the distribution of immunoglobulin gene subgroups present in the four libraries has revealed shifts in the B cell repertoire originating from differences in genetic background and immunological status of mice.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Liu, Guoxia; Arnaud, Philippe; Offmann, Bernard; Picimbon, Jean-François
Pheromone, Natural Odor and Odorant Reception Suppressing Agent (ORSA) for Insect Control Book Section
Dans: Olfactory Concepts of Insect Control-Alternative to Insecticides, p. 311–345, Springer, Cham, 2019.
@incollection{liu2019pheromone,
title = {Pheromone, Natural Odor and Odorant Reception Suppressing Agent (ORSA) for Insect Control},
author = {Guoxia Liu and Philippe Arnaud and Bernard Offmann and Jean-François Picimbon},
doi = {10.1007/978-3-030-05165-5},
year = {2019},
date = {2019-01-01},
booktitle = {Olfactory Concepts of Insect Control-Alternative to Insecticides},
pages = {311--345},
publisher = {Springer, Cham},
abstract = {Odorant-binding proteins (OBPs) are small ``bowl-like'' globular pro- teins, highly abundant in the antennae of most insect species. These proteins are believed to mediate reception of odor molecules at the periphery of sensory receptor neurons. Therefore, they may represent crucial targets for becoming new methods of insect pest control by directly interfering with the olfactory acuity of the insect. The current better understanding of molecular mechanisms underlying odor detec- tion and the knowledge about the functional binding sites of OBPs and many other families of binding proteins in various insect species is elucidated here. Such infor- mation forms the basis for the synthesis of new inhibitor olfactory compounds (Odorant Reception-Suppressing Agents, ORSAs) to interact specifically with the groups of insect pests.},
keywords = {},
pubstate = {published},
tppubtype = {incollection}
}
Vetrivel, Iyanar; Hoffmann, Lionel; Guegan, Sean; Offmann, Bernard; Laurent, Adele D
PBmapclust: Mapping and Clustering the Protein Conformational Space Using a Structural Alphabet Proceedings Article
Dans: Byska, Jan; Krone, Michael; Sommer, Björn (Ed.): Workshop on Molecular Graphics and Visual Analysis of Molecular Data, The Eurographics Association, 2019, ISBN: 978-3-03868-085-7.
@inproceedings{lva.20191097b,
title = {PBmapclust: Mapping and Clustering the Protein Conformational Space Using a Structural Alphabet},
author = {Iyanar Vetrivel and Lionel Hoffmann and Sean Guegan and Bernard Offmann and Adele D Laurent},
editor = {Jan Byska and Michael Krone and Björn Sommer},
doi = {10.2312/molva.20191097},
isbn = {978-3-03868-085-7},
year = {2019},
date = {2019-01-01},
booktitle = {Workshop on Molecular Graphics and Visual Analysis of Molecular Data},
publisher = {The Eurographics Association},
abstract = {Analyzing the data from molecular dynamics simulation of biological macromolecules like proteins is challenging. We propose a simple tool called PBmapclust that is based on a well established structural alphabet called Protein blocks (PB). PBs help in tracing the trajectory of the protein backbone by categorizing it into 16 distinct structural states. PBmapclust provides a time vs. amino acid residue plot that is color coded to match each of the PBs. Color changes correspond to structural changes, giving a visual overview of the simulation. Further, PBmapclust enables the user to "map" the conformational space sampled by the protein during the MD simulation by clustering the conformations. The ability to generate sub-maps for specific residues and specific time intervals allows the user to focus on residues of interest like for active sites or disordered regions. We have included an illustrative case study to demonstrate the utility of the tool. It describes the effect of the disordered domain of a HSP90 co-chaperone on the conformation of its active site residues. The scripts required to perform PBmapclust are made freely available under the GNU general public license.},
keywords = {},
pubstate = {published},
tppubtype = {inproceedings}
}
Vetrivel, Iyanar; de Brevern, Alexandre G; Cadet, Frédéric; Srinivasan, Narayanaswamy; Offmann, Bernard
Structural variations within proteins can be as large as variations observed across their homologues Article de journal
Dans: Biochimie, vol. 167, p. 162–170, 2019, ISSN: 61831638.
@article{Vetrivel2019,
title = {Structural variations within proteins can be as large as variations observed across their homologues},
author = {Iyanar Vetrivel and Alexandre G de Brevern and Frédéric Cadet and Narayanaswamy Srinivasan and Bernard Offmann},
doi = {10.1016/j.biochi.2019.09.013},
issn = {61831638},
year = {2019},
date = {2019-01-01},
journal = {Biochimie},
volume = {167},
pages = {162--170},
abstract = {Understanding the structural plasticity of proteins is key to understanding the intricacies of their functions and mechanistic basis. In the current study, we analyzed the available multiple crystal structures of the same protein for the structural differences. For this purpose we used an abstraction of protein structures referred as Protein Blocks (PBs) that was previously established. We also characterized the nature of the structural variations for a few proteins using molecular dynamics simulations. In both the cases, the structural variations were summarized in the form of substitution matrices of PBs. We show that certain conformational states are preferably replaced by other specific conformational states. Interestingly, these structural variations are highly similar to those previously observed across structures of homologous proteins (r2 = 0.923) or across the ensemble of conformations from NMR data (r2 = 0.919). Thus our study quantitatively shows that overall trends of structural changes in a given protein are nearly identical to the trends of structural differences that occur in the topologically equivalent positions in homologous proteins. Specific case studies are used to illustrate the nature of these structural variations.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
1 publication
Cadet, Frédéric; Fontaine, Nicolas; Li, Guangyue; Sanchis, Joaquin; Chong, Matthieu Ng Fuk; Pandjaitan, Rudy; Vetrivel, Iyanar; Offmann, Bernard; Reetz, Manfred T
A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes Article de journal
Dans: Scientific Reports, vol. 8, no. 1, p. 1–15, 2018, ISSN: 20452322.
@article{Cadet2018,
title = {A machine learning approach for reliable prediction of amino acid interactions and its application in the directed evolution of enantioselective enzymes},
author = {Frédéric Cadet and Nicolas Fontaine and Guangyue Li and Joaquin Sanchis and Matthieu {Ng Fuk Chong} and Rudy Pandjaitan and Iyanar Vetrivel and Bernard Offmann and Manfred T Reetz},
doi = {10.1038/s41598-018-35033-y},
issn = {20452322},
year = {2018},
date = {2018-01-01},
journal = {Scientific Reports},
volume = {8},
number = {1},
pages = {1--15},
abstract = {Directed evolution is an important research activity in synthetic biology and biotechnology. Numerous reports describe the application of tedious mutation/screening cycles for the improvement of proteins. Recently, knowledge-based approaches have facilitated the prediction of protein properties and the identification of improved mutants. However, epistatic phenomena constitute an obstacle which can impair the predictions in protein engineering. We present an innovative sequence-activity relationship (innov'SAR) methodology based on digital signal processing combining wet-lab experimentation and computational protein design. In our machine learning approach, a predictive model is developed to find the resulting property of the protein when the n single point mutations are permuted (2n combinations). The originality of our approach is that only sequence information and the fitness of mutants measured in the wet-lab are needed to build models. We illustrate the application of the approach in the case of improving the enantioselectivity of an epoxide hydrolase from Aspergillus niger. n = 9 single point mutants of the enzyme were experimentally assessed for their enantioselectivity and used as a learning dataset to build a model. Based on combinations of the 9 single point mutations (29), the enantioselectivity of these 512 variants were predicted, and candidates were experimentally checked: better mutants with higher enantioselectivity were indeed found.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
3 publications
Vetrivel, Iyanar; Mahajan, Swapnil; Tyagi, Manoj; Hoffmann, Lionel; Sanejouand, Yves-Henri; Srinivasan, Narayanaswamy; Brevern, Alexandre G De; Cadet, Frédéric; Offmann, Bernard
Knowledge-based prediction of protein backbone conformation using a structural alphabet Article de journal
Dans: PLoS ONE, vol. 12, no. 11, 2017, ISSN: 19326203.
@article{Vetrivel2017,
title = {Knowledge-based prediction of protein backbone conformation using a structural alphabet},
author = {Iyanar Vetrivel and Swapnil Mahajan and Manoj Tyagi and Lionel Hoffmann and Yves-Henri Sanejouand and Narayanaswamy Srinivasan and Alexandre G {De Brevern} and Frédéric Cadet and Bernard Offmann},
doi = {10.1371/journal.pone.0186215},
issn = {19326203},
year = {2017},
date = {2017-11-01},
journal = {PLoS ONE},
volume = {12},
number = {11},
publisher = {Public Library of Science},
abstract = {Libraries of structural prototypes that abstract protein local structures are known as structural alphabets and have proven to be very useful in various aspects of protein structure analyses and predictions. One such library, Protein Blocks, is composed of 16 standard 5-residues long structural prototypes. This form of analyzing proteins involves drafting its structure as a string of Protein Blocks. Predicting the local structure of a protein in terms of protein blocks is the general objective of this work. A new approach, PB-kPRED is proposed towards this aim. It involves (i) organizing the structural knowledge in the form of a database of pentapeptide fragments extracted from all protein structures in the PDB and (ii) applying a knowledge-based algorithm that does not rely on any secondary structure predictions and/ or sequence alignment profiles, to scan this database and predict most probable backbone conformations for the protein local structures. Though PB-kPRED uses the structural information from homologues in preference, if available. The predictions were evaluated rigorously on 15,544 query proteins representing a non-redundant subset of the PDB filtered at 30% sequence identity cut-off. We have shown that the kPRED method was able to achieve mean accuracies ranging from 40.8% to 66.3% depending on the availability of homologues. The impact of the different strategies for scanning the database on the prediction was evaluated and is discussed. Our results highlight the usefulness of the method in the context of proteins without any known structural homologues. A scoring function that gives a good estimate of the accuracy of prediction was further developed. This score estimates very well the accuracy of the algorithm (R2 of 0.82). An online version of the tool is provided freely for non-commercial usage at http://www.bo-protscience.fr/kpred/.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Labbé, Pauline; Faure, Emilie; Lecointe, Simon; Scouarnec, Solena Le; Kyndt, Florence; Marrec, Marie; Tourneau, Thierry Le; Offmann, Bernard; Duplaà, Cécile; Zaffran, Stéphane; Schott, Jean Jacques; Merot, Jean
The alternatively spliced LRRFIP1 Isoform-1 is a key regulator of the Wnt/β-catenin transcription pathway Article de journal
Dans: Biochimica et Biophysica Acta - Molecular Cell Research, vol. 1864, no. 7, p. 1142–1152, 2017, ISSN: 18792596.
@article{Labbe2017,
title = {The alternatively spliced LRRFIP1 Isoform-1 is a key regulator of the Wnt/β-catenin transcription pathway},
author = {Pauline Labbé and Emilie Faure and Simon Lecointe and Solena {Le Scouarnec} and Florence Kyndt and Marie Marrec and Thierry {Le Tourneau} and Bernard Offmann and Cécile Duplaà and Stéphane Zaffran and Jean Jacques Schott and Jean Merot},
doi = {10.1016/j.bbamcr.2017.03.008},
issn = {18792596},
year = {2017},
date = {2017-01-01},
journal = {Biochimica et Biophysica Acta - Molecular Cell Research},
volume = {1864},
number = {7},
pages = {1142--1152},
abstract = {The GC-rich Binding Factor 2/Leucine Rich Repeat in the Flightless 1 Interaction Protein 1 gene (GCF2/LRRFIP1) is predicted to be alternatively spliced in five different isoforms. Although important peptide sequence differences are expected to result from this alternative splicing, to date, only the gene transcription regulator properties of LRRFIP1-Iso5 were unveiled. Based on molecular, cellular and biochemical data, we show here that the five isoforms define two molecular entities with different expression profiles in human tissues, subcellular localizations, oligomerization properties and transcription enhancer properties of the canonical Wnt pathway. We demonstrated that LRRFIP1-Iso3, -4 and -5, which share over 80% sequence identity, are primarily located in the cell cytoplasm and form homo and hetero-multimers between each other. In contrast, LRRFIP1-Iso1 and -2 are primarily located in the cell nucleus in part thanks to their shared C-terminal domain. Furthermore, we showed that LRRFIP1-Iso1 is preferentially expressed in the myocardium and skeletal muscle. Using the in vitro Topflash reporter assay we revealed that among LRRFIP1 isoforms, LRRFIP1-Iso1 is the strongest enhancer of the β-catenin Wnt canonical transcription pathway thanks to a specific N-terminal domain harboring two critical tryptophan residues (W76, 82). In addition, we showed that the Wnt enhancer properties of LRRFIP1-Iso1 depend on its homo-dimerisation which is governed by its specific coiled coil domain. Together our study identified LRRFIP1-Iso1 as a critical regulator of the Wnt canonical pathway with a potential role in myocyte differentiation and myogenesis.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Shahsavarian, Melody A; Chaaya, Nancy; Costa, Narciso; Boquet, Didier; Atkinson, Alexandre; Offmann, Bernard; Kaveri, Srini V; Lacroix-Desmazes, Sébastien; Friboulet, Alain; Avalle, Bérangère; Padiolleau-Lefèvre, Séverine
Multitarget selection of catalytic antibodies with β-lactamase activity using phage display Article de journal
Dans: FEBS Journal, vol. 284, no. 4, p. 634–653, 2017, ISSN: 17424658.
@article{Shahsavarian2017,
title = {Multitarget selection of catalytic antibodies with β-lactamase activity using phage display},
author = {Melody A Shahsavarian and Nancy Chaaya and Narciso Costa and Didier Boquet and Alexandre Atkinson and Bernard Offmann and Srini V Kaveri and Sébastien Lacroix-Desmazes and Alain Friboulet and Bérangère Avalle and Séverine Padiolleau-Lefèvre},
doi = {10.1111/febs.14012},
issn = {17424658},
year = {2017},
date = {2017-01-01},
journal = {FEBS Journal},
volume = {284},
number = {4},
pages = {634--653},
abstract = {β-lactamase enzymes responsible for bacterial resistance to antibiotics are among the most important health threats to the human population today. Understanding the increasingly vast structural motifs responsible for the catalytic mechanism of β-lactamases will help improve the future design of new generation antibiotics and mechanism-based inhibitors of these enzymes. Here we report the construction of a large murine single chain fragment variable (scFv) phage display library of size 2.7 × 109 with extended diversity by combining different mouse models. We have used two molecularly different inhibitors of the R-TEM β-lactamase as targets for selection of catalytic antibodies with β-lactamase activity. This novel methodology has led to the isolation of five antibody fragments, which are all capable of hydrolyzing the β-lactam ring. Structural modeling of the selected scFv has revealed the presence of different motifs in each of the antibody fragments potentially responsible for their catalytic activity. Our results confirm (a) the validity of using our two target inhibitors for the in vitro selection of catalytic antibodies endowed with β-lactamase activity, and (b) the plasticity of the β-lactamase active site responsible for the wide resistance of these enzymes to clinically available inhibitors and antibiotics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2 publications
Liu, Guoxia; Ma, Hongmei; Xie, Hongyan; Xuan, Ning; Guo, Xia; Fan, Zhongxue; Rajashekar, Balaji; Arnaud, Philippe; Offmann, Bernard; Picimbon, Jean François
Biotype characterization, developmental profiling, insecticide response and binding property of Bemisia tabaci chemosensory proteins: Role of CSP in insect defense Article de journal
Dans: PLoS ONE, vol. 11, no. 5, 2016, ISSN: 19326203.
@article{Liu2016,
title = {Biotype characterization, developmental profiling, insecticide response and binding property of Bemisia tabaci chemosensory proteins: Role of CSP in insect defense},
author = {Guoxia Liu and Hongmei Ma and Hongyan Xie and Ning Xuan and Xia Guo and Zhongxue Fan and Balaji Rajashekar and Philippe Arnaud and Bernard Offmann and Jean François Picimbon},
doi = {10.1371/journal.pone.0154706},
issn = {19326203},
year = {2016},
date = {2016-05-01},
journal = {PLoS ONE},
volume = {11},
number = {5},
publisher = {Public Library of Science},
abstract = {Chemosensory proteins (CSPs) are believed to play a key role in the chemosensory process in insects. Sequencing genomic DNA and RNA encoding CSP1, CSP2 and CSP3 in the sweet potato whitefly Bemisia tabaci showed strong variation between B and Q biotypes. Analyzing CSP-RNA levels showed not only biotype, but also age and developmental stage-specific expression. Interestingly, applying neonicotinoid thiamethoxam insecticide using twenty-five different dose/time treatments in B and Q young adults showed that Bemisia CSP1, CSP2 and CSP3 were also differentially regulated over insecticide exposure. In our study one of the adult-specific gene (CSP1) was shown to be significantly up-regulated by the insecticide in Q, the most highly resistant form of B. tabaci. Correlatively, competitive binding assays using tryptophan fluorescence spectroscopy and molecular docking demonstrated that CSP1 protein preferentially bound to linoleic acid, while CSP2 and CSP3 proteins rather associated to another completely different type of chemical, i.e. α-pentyl-cinnamaldehyde (jasminaldehyde). This might indicate that some CSPs in whiteflies are crucial to facilitate the transport of fatty acids thus regulating some metabolic pathways of the insect immune response, while some others are tuned to much more volatile chemicals known not only for their pleasant odor scent, but also for their potent toxic insecticide activity.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Verhaeghe, Tom; Winter, Karel De; Berland, Magali; Vreese, Rob De; D'Hooghe, Matthias; Offmann, Bernard; Desmet, Tom
Converting bulk sugars into prebiotics: Semi-rational design of a transglucosylase with controlled selectivity Article de journal
Dans: Chemical Communications, vol. 52, no. 18, p. 3687–3689, 2016, ISSN: 1364548X.
@article{Verhaeghe2016,
title = {Converting bulk sugars into prebiotics: Semi-rational design of a transglucosylase with controlled selectivity},
author = {Tom Verhaeghe and Karel {De Winter} and Magali Berland and Rob {De Vreese} and Matthias D'Hooghe and Bernard Offmann and Tom Desmet},
doi = {10.1039/c5cc09940d},
issn = {1364548X},
year = {2016},
date = {2016-01-01},
journal = {Chemical Communications},
volume = {52},
number = {18},
pages = {3687--3689},
publisher = {Royal Society of Chemistry},
abstract = {Despite the growing importance of prebiotics in nutrition and gastroenterology, their structural variety is currently still very limited. The lack of straightforward procedures to gain new products in sufficient amounts often hampers application testing and further development. Although the enzyme sucrose phosphorylase can be used to produce the rare disaccharide kojibiose (α-1,2-glucobiose) from the bulk sugars sucrose and glucose, the target compound is only a side product that is difficult to isolate. Accordingly, for this biocatalyst to become economically attractive, the formation of other glucobioses should be avoided and therefore we applied semi-rational mutagenesis and low-throughput screening, which resulted in a double mutant (L341I-Q345S) with a selectivity of 95% for kojibiose. That way, an efficient and scalable production process with a yield of 74% could be established, and with a simple yeast treatment and crystallization step over a hundred grams of highly pure kojibiose (textgreater99.5%) was obtained.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2 publications
Mahajan, Swapnil; Brevern, Alexandre G De; Sanejouand, Yves-Henri; Srinivasan, Narayanaswamy; Offmann, Bernard
Use of a structural alphabet to find compatible folds for amino acid sequences Article de journal
Dans: Protein Science, vol. 24, no. 1, p. 145–153, 2015, ISSN: 1469896X.
@article{Mahajan2015a,
title = {Use of a structural alphabet to find compatible folds for amino acid sequences},
author = {Swapnil Mahajan and Alexandre G {De Brevern} and Yves-Henri Sanejouand and Narayanaswamy Srinivasan and Bernard Offmann},
doi = {10.1002/pro.2581},
issn = {1469896X},
year = {2015},
date = {2015-01-01},
journal = {Protein Science},
volume = {24},
number = {1},
pages = {145--153},
abstract = {The structural annotation of proteins with no detectable homologs of known 3D structure identified using sequence-search methods is a major challenge today. We propose an original method that computes the conditional probabilities for the amino-acid sequence of a protein to fit to known protein 3D structures using a structural alphabet, known as "Protein Blocks" (PBs). PBs constitute a library of 16 local structural prototypes that approximate every part of protein backbone structures. It is used to encode 3D protein structures into 1D PB sequences and to capture sequence to structure relationships. Our method relies on amino acid occurrence matrices, one for each PB, to score global and local threading of query amino acid sequences to protein folds encoded into PB sequences. It does not use any information from residue contacts or sequence-search methods or explicit incorporation of hydrophobic effect. The performance of the method was assessed with independent test datasets derived from SCOP 1.75A. With a Z-score cutoff that achieved 95% specificity (i.e., less than 5% false positives), global and local threading showed sensitivity of 64.1% and 34.2%, respectively. We further tested its performance on 57 difficult CASP10 targets that had no known homologs in PDB: 38 compatible templates were identified by our approach and 66% of these hits yielded correctly predicted structures. This method scales-up well and offers promising perspectives for structural annotations at genomic level. It has been implemented in the form of a web-server that is freely available at http://www.bo-protscience.fr/forsa.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fontaine, Nicolas; Grondin-perez, Brigitte; Cadet, Frédéric; Offmann, Bernard; Fontaine, Nicolas; Grondin-perez, Brigitte; Cadet, Frédéric; Offmann, Bernard; Fontaine, Nicolas; Grondin-perez, Brigitte; Cadet, Frédéric; Offmann, Bernard
Modeling of a Cell-Free Synthetic System for Biohydrogen Production Article de journal
Dans: Journal of Computer Science & Systems Biology, vol. 8, no. 3, 2015, ISSN: 09747230.
@article{Fontaine2015,
title = {Modeling of a Cell-Free Synthetic System for Biohydrogen Production},
author = {Nicolas Fontaine and Brigitte Grondin-perez and Frédéric Cadet and Bernard Offmann and Nicolas Fontaine and Brigitte Grondin-perez and Frédéric Cadet and Bernard Offmann and Nicolas Fontaine and Brigitte Grondin-perez and Frédéric Cadet and Bernard Offmann},
doi = {10.4172/jcsb.1000181},
issn = {09747230},
year = {2015},
date = {2015-01-01},
journal = {Journal of Computer Science & Systems Biology},
volume = {8},
number = {3},
abstract = {Hydrogen is a good candidate for the next generation fuel with a high energy density and an environment friendly behavior in the energy production phase. Micro-organism based biological production of hydrogen currently suffers low hydrogen production yields because the living cells must sustain different cellular activities other than the hydrogen production to survive. To circumvent this, teams have explored the synthetic assembly of enzymes in-vitro in cell-free systems with specific functions. Such a synthetic cell-free system was recently devised by combining 13 different enzymes to synthesize hydrogen from cellulose or cellobiose with better yield than microorganism-based systems. We used methods based on differential equations calculations to investigate how the initial conditions and the kinetic parameters of the enzymes influenced the productivity of a such system and, through simulations, identified those conditions that would optimize hydrogen production starting with cellobiose as substrate. Further, if the kinetic parameters of the component enzymes of such a system are not known, we showed how, using artificial neural network, it is possible to identify alternative models that account for the rate of production of hydrogen. This work demonstrates how modeling can help in designing and characterizing cell-free systems in synthetic biology. A web-based simulator implementing our differential equations based model is provided freely as a service for non- commercial usage at http://www.bo-protscience.fr/h2.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2014
Berland, Magali; Offmann, Bernard; André, Isabelle; Remaud-Siméon, Magali; Charton, Philippe
A web-based tool for rational screening of mutants libraries using ProSAR Article de journal
Dans: Protein Eng Des Sel, vol. 27, no. 10, p. 375-81, 2014.
@article{Berland:2014aa,
title = {A web-based tool for rational screening of mutants libraries using ProSAR},
author = {Magali Berland and Bernard Offmann and Isabelle André and Magali Remaud-Siméon and Philippe Charton},
doi = {10.1093/protein/gzu035},
year = {2014},
date = {2014-10-01},
urldate = {2014-10-01},
journal = {Protein Eng Des Sel},
volume = {27},
number = {10},
pages = {375-81},
abstract = {In directed evolution experiments, it is at stake to have methods to screen efficiently the mutant libraries. We propose a web-based tool that implements an established in silico method for the rational screening of mutant libraries. The method, known as ProSAR, attempts to link sequence data to activity. The method uses statistical models trained on small experimental datasets provided by the user. These can integrate potential epistatic interactions between mutations and be used in many diverse biological contexts. It drastically improves the search for leading mutants. The tool is freely available to non-commercial users at http://bo-protscience.fr/prosar/.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Mahajan, Swapnil; de Brevern, Alexandre G; Offmann, Bernard; Srinivasan, Narayanaswamy
Correlation between local structural dynamics of proteins inferred from NMR ensembles and evolutionary dynamics of homologues of known structure Article de journal
Dans: J Biomol Struct Dyn, vol. 32, no. 5, p. 751-8, 2014.
@article{Mahajan:2014aa,
title = {Correlation between local structural dynamics of proteins inferred from NMR ensembles and evolutionary dynamics of homologues of known structure},
author = {Swapnil Mahajan and Alexandre G {de Brevern} and Bernard Offmann and Narayanaswamy Srinivasan},
doi = {10.1080/07391102.2013.789989},
year = {2014},
date = {2014-01-01},
journal = {J Biomol Struct Dyn},
volume = {32},
number = {5},
pages = {751-8},
abstract = {Conformational changes in proteins are extremely important for their biochemical functions. Correlation between inherent conformational variations in a protein and conformational differences in its homologues of known structure is still unclear. In this study, we have used a structural alphabet called Protein Blocks (PBs). PBs are used to perform abstraction of protein 3-D structures into a 1-D strings of 16 alphabets (a-p) based on dihedral angles of overlapping pentapeptides. We have analyzed the variations in local conformations in terms of PBs represented in the ensembles of 801 protein structures determined using NMR spectroscopy. In the analysis of concatenated data over all the residues in all the NMR ensembles, we observe that the overall nature of inherent local structural variations in NMR ensembles is similar to the nature of local structural differences in homologous proteins with a high correlation coefficient of .94. High correlation at the alignment positions corresponding to helical and β-sheet regions is only expected. However, the correlation coefficient by considering only the loop regions is also quite high (.91). Surprisingly, segregated position-wise analysis shows that this high correlation does not hold true to loop regions at the structurally equivalent positions in NMR ensembles and their homologues of known structure. This suggests that the general nature of local structural changes is unique; however most of the local structural variations in loop regions of NMR ensembles do not correlate to their local structural differences at structurally equivalent positions in homologues.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Manoharan, Malini; Fuchs, Patrick F J; Sowdhamini, Ramanathan; Offmann, Bernard
Insights on pH-dependent conformational changes of mosquito odorant binding proteins by molecular dynamics simulations Article de journal
Dans: J Biomol Struct Dyn, vol. 32, no. 11, p. 1742-51, 2014.
@article{Manoharan:2014aa,
title = {Insights on pH-dependent conformational changes of mosquito odorant binding proteins by molecular dynamics simulations},
author = {Malini Manoharan and Patrick F J Fuchs and Ramanathan Sowdhamini and Bernard Offmann},
doi = {10.1080/07391102.2013.834118},
year = {2014},
date = {2014-01-01},
journal = {J Biomol Struct Dyn},
volume = {32},
number = {11},
pages = {1742-51},
abstract = {Chemical recognition plays an important role for the survival and reproduction of many insect species. Odorant binding proteins (OBPs) are the primary components of the insect olfactory mechanism and have been documented to play an important role in the host-seeking mechanism of mosquitoes. They are "transport proteins" believed to transport odorant molecules from the external environment to their respective membrane targets, the olfactory receptors. The mechanism by which this transport occurs in mosquitoes remains a conundrum in this field. Nevertheless, OBPs have proved to be amenable to conformational changes mediated by a pH change in other insect species. In this paper, the effect of pH on the conformational flexibility of mosquito OBPs is assessed computationally using molecular dynamics simulations of a mosquito OBP "CquiOBP1" bound to its pheromone 3OG (PDB ID: 3OGN). Conformational twist of a loop, driven by a set of well-characterized changes in intramolecular interactions of the loop, is demonstrated. The concomitant (i) closure of what is believed to be the entrance of the binding pocket, (ii) expansion of what could be an exit site, and (iii) migration of the ligand towards this putative exit site provide preliminary insights into the mechanism of ligand binding and release of these proteins in mosquitoes. The correlation of our results with previous experimental observations based on NMR studies help us provide a cardinal illustration on one of the probable dynamics and mechanism by which certain mosquito OBPs could deliver their ligand to their membrane-bound receptors at specific pH conditions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2013
Mahajan, Swapnil; Agarwal, Garima; Iftekhar, Mohammed; Offmann, Bernard; de Brevern, Alexandre G; Srinivasan, Narayanaswamy
DoSA: Database of Structural Alignments Article de journal
Dans: Database (Oxford), vol. 2013, p. bat048, 2013.
@article{Mahajan:2013aa,
title = {DoSA: Database of Structural Alignments},
author = {Swapnil Mahajan and Garima Agarwal and Mohammed Iftekhar and Bernard Offmann and Alexandre G {de Brevern} and Narayanaswamy Srinivasan},
doi = {10.1093/database/bat048},
year = {2013},
date = {2013-01-01},
journal = {Database (Oxford)},
volume = {2013},
pages = {bat048},
abstract = {Protein structure alignment is a crucial step in protein structure-function analysis. Despite the advances in protein structure alignment algorithms, some of the local conformationally similar regions are mislabeled as structurally variable regions (SVRs). These regions are not well superimposed because of differences in their spatial orientations. The Database of Structural Alignments (DoSA) addresses this gap in identification of local structural similarities obscured in global protein structural alignments by realigning SVRs using an algorithm based on protein blocks. A set of protein blocks is a structural alphabet that abstracts protein structures into 16 unique local structural motifs. DoSA provides unique information about 159,780 conformationally similar and 56,140 conformationally dissimilar SVRs in 74 705 pairwise structural alignments of homologous proteins. The information provided on conformationally similar and dissimilar SVRs can be helpful to model loop regions. It is also conceivable that conformationally similar SVRs with conserved residues could potentially contribute toward functional integrity of homologues, and hence identifying such SVRs could be helpful in understanding the structural basis of protein function. Database URL: http://bo-protscience.fr/dosa/},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Manoharan, Malini; Chong, Matthieu Ng Fuk; Vaïtinadapoulé, Aurore; Frumence, Etienne; Sowdhamini, Ramanathan; Offmann, Bernard
Comparative genomics of odorant binding proteins in Anopheles gambiae, Aedes aegypti, and Culex quinquefasciatus Article de journal
Dans: Genome Biol Evol, vol. 5, no. 1, p. 163-80, 2013.
@article{Manoharan:2013ab,
title = {Comparative genomics of odorant binding proteins in Anopheles gambiae, Aedes aegypti, and Culex quinquefasciatus},
author = {Malini Manoharan and Matthieu Ng Fuk Chong and Aurore Vaïtinadapoulé and Etienne Frumence and Ramanathan Sowdhamini and Bernard Offmann},
doi = {10.1093/gbe/evs131},
year = {2013},
date = {2013-01-01},
journal = {Genome Biol Evol},
volume = {5},
number = {1},
pages = {163-80},
abstract = {About 1 million people in the world die each year from diseases spread by mosquitoes, and understanding the mechanism of host identification by the mosquitoes through olfaction is at stake. The role of odorant binding proteins (OBPs) in the primary molecular events of olfaction in mosquitoes is becoming an important focus of biological research in this area. Here, we present a comprehensive comparative genomics study of OBPs in the three disease-transmitting mosquito species Anopheles gambiae, Aedes aegypti, and Culex quinquefasciatus starting with the identification of 110 new OBPs in these three genomes. We have characterized their genomic distribution and orthologous and phylogenetic relationships. The diversity and expansion observed with respect to the Aedes and Culex genomes suggests that the OBP gene family acquired functional diversity concurrently with functional constraints posed on these two species. Sequences with unique features have been characterized such as the "two-domain OBPs" (previously known as Atypical OBPs) and "MinusC OBPs" in mosquito genomes. The extensive comparative genomics featured in this work hence provides useful primary insights into the role of OBPs in the molecular adaptations of mosquito olfactory system and could provide more clues for the identification of potential targets for insect repellants and attractants.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Manoharan, Malini; Sankar, Kannan; Offmann, Bernard; Ramanathan, Sowdhamini
Association of putative members to family of mosquito odorant binding proteins: scoring scheme using fuzzy functional templates and cys residue positions Article de journal
Dans: Bioinform Biol Insights, vol. 7, p. 231-51, 2013.
@article{Manoharan:2013aa,
title = {Association of putative members to family of mosquito odorant binding proteins: scoring scheme using fuzzy functional templates and cys residue positions},
author = {Malini Manoharan and Kannan Sankar and Bernard Offmann and Sowdhamini Ramanathan},
doi = {10.4137/BBI.S11096},
year = {2013},
date = {2013-01-01},
journal = {Bioinform Biol Insights},
volume = {7},
pages = {231-51},
abstract = {Proteins may be related to each other very specifically as homologous subfamilies. Proteins can also be related to diverse proteins at the super family level. It has become highly important to characterize the existing sequence databases by their signatures to facilitate the function annotation of newly added sequences. The algorithm described here uses a scheme for the classification of odorant binding proteins on the basis of functional residues and Cys-pairing. The cysteine-based scoring scheme not only helps in unambiguously identifying families like odorant binding proteins (OBPs), but also aids in their classification at the subfamily level with reliable accuracy. The algorithm was also applied to yet another cysteine-rich family, where similar accuracy was observed that ensures the application of the protocol to other families.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2011
Chilamakuri, Chandra Sekhar Reddy; Joshi, Adwait; Rani, Sane Sudha; Offmann, Bernard; Sowdhamini, R
Cross-Genome Comparisons of Newly Identified Domains in Mycoplasma gallisepticum and Domain Architectures with Other Mycoplasma species Article de journal
Dans: Comparative and Functional Genomics, vol. 2011, 2011.
@article{Chilamakuri:2011aa,
title = {Cross-Genome Comparisons of Newly Identified Domains in Mycoplasma gallisepticum and Domain Architectures with Other Mycoplasma species},
author = {Chandra Sekhar Reddy Chilamakuri and Adwait Joshi and Sane Sudha Rani and Bernard Offmann and R Sowdhamini},
doi = {10.1155/2011/878973},
year = {2011},
date = {2011-01-01},
journal = {Comparative and Functional Genomics},
volume = {2011},
abstract = {Accurate functional annotation of protein sequences is hampered by important factors such as the failure of sequence search methods to identify relationships and the inherent diversity in function of proteins related at low sequence similarities. Earlier, we had employed intermediate sequence search approach to establish new domain relationships in the unassigned regions of gene products at the whole genome level by taking Mycoplasma gallisepticum as a specific example and established new domain relationships. In this paper, we report a detailed comparison of the conservation status of the domain and domain architectures of the gene products that bear our newly predicted domains amongst 14 other Mycoplasma genomes and reported the probable implications for the organisms. Some of the domain associations, observed in Mycoplasma that afflict humans and other non-human primates, are involved in regulation of solute transport and DNA binding suggesting specific modes of host-pathogen interactions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2010
Joseph, Agnel Praveen; Agarwal, Garima; Mahajan, Swapnil; Gelly, Jean-Christophe; Swapna, Lakshmipuram S; Offmann, Bernard; Cadet, Frédéric; Bornot, Aurélie; Tyagi, Manoj; Valadié, Hélène; Schneider, Bohdan; Etchebest, Catherine; Srinivasan, Narayanaswamy; de Brevern, Alexandre G
A short survey on protein blocks Article de journal
Dans: Biophys Rev, vol. 2, no. 3, p. 137-147, 2010.
@article{Joseph:2010aa,
title = {A short survey on protein blocks},
author = {Agnel Praveen Joseph and Garima Agarwal and Swapnil Mahajan and Jean-Christophe Gelly and Lakshmipuram S Swapna and Bernard Offmann and Frédéric Cadet and Aurélie Bornot and Manoj Tyagi and Hélène Valadié and Bohdan Schneider and Catherine Etchebest and Narayanaswamy Srinivasan and Alexandre G {de Brevern}},
doi = {10.1007/s12551-010-0036-1},
year = {2010},
date = {2010-08-01},
journal = {Biophys Rev},
volume = {2},
number = {3},
pages = {137-147},
abstract = {Protein structures are classically described in terms of secondary structures. Even if the regular secondary structures have relevant physical meaning, their recognition from atomic coordinates has some important limitations such as uncertainties in the assignment of boundaries of helical and β-strand regions. Further, on an average about 50% of all residues are assigned to an irregular state, i.e., the coil. Thus different research teams have focused on abstracting conformation of protein backbone in the localized short stretches. Using different geometric measures, local stretches in protein structures are clustered in a chosen number of states. A prototype representative of the local structures in each cluster is generally defined. These libraries of local structures prototypes are named as "structural alphabets". We have developed a structural alphabet, named Protein Blocks, not only to approximate the protein structure, but also to predict them from sequence. Since its development, we and other teams have explored numerous new research fields using this structural alphabet. We review here some of the most interesting applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Reddy, Chilamakuri Cs; Rani, Sane Sudha; Offmann, Bernard; Sowdhamini, R
Systematic search for putative new domain families in Mycoplasma gallisepticum genome Article de journal
Dans: BMC Res Notes, vol. 3, p. 98, 2010.
@article{Reddy:2010aa,
title = {Systematic search for putative new domain families in Mycoplasma gallisepticum genome},
author = {Chilamakuri Cs Reddy and Sane Sudha Rani and Bernard Offmann and R Sowdhamini},
doi = {10.1186/1756-0500-3-98},
year = {2010},
date = {2010-04-01},
journal = {BMC Res Notes},
volume = {3},
pages = {98},
abstract = {BACKGROUND: Protein domains are the fundamental units of protein structure, function and evolution. The delineation of different domains in proteins is important for classification, understanding of structure, function and evolution. The delineation of protein domains within a polypeptide chain, namely at the genome scale, can be achieved in several ways but may remain problematic in many instances. Difficulties in identifying the domain content of a given sequence arise when the query sequence has no homologues with experimentally determined structure and searching against sequence domain databases also results in insignificant matches. Identification of domains under low sequence identity conditions and lack of structural homologues acquire a crucial importance especially at the genomic scale. FINDINGS: We have developed a new method for the identification of domains in unassigned regions through indirect connections and scaled up its application to the analysis of 434 unassigned regions in 726 protein sequences of Mycoplasma gallisepticum genome. We could establish 71 new domain relationships and probable 63 putative new domain families through intermediate sequences in the unassigned regions, which importantly represent an overall 10% increase in PfamA domain annotation over the direct assignment in this genome. CONCLUSIONS: The systematic analysis of the unassigned regions in the Mycoplasma gallisepticum genome has provided some insight into the possible new domain relationships and putative new domain families. Further investigation of these predicted new domains may prove beneficial in improving the existing domain prediction algorithms.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Groben, René; Kaloudas, Dimitrios; Raines, Christine A; Offmann, Bernard; Maberly, Stephen C; Gontero, Brigitte
Comparative sequence analysis of CP12, a small protein involved in the formation of a Calvin cycle complex in photosynthetic organisms Article de journal
Dans: Photosynth Res, vol. 103, no. 3, p. 183-94, 2010.
@article{Groben:2010aa,
title = {Comparative sequence analysis of CP12, a small protein involved in the formation of a Calvin cycle complex in photosynthetic organisms},
author = {René Groben and Dimitrios Kaloudas and Christine A Raines and Bernard Offmann and Stephen C Maberly and Brigitte Gontero},
doi = {10.1007/s11120-010-9542-z},
year = {2010},
date = {2010-03-01},
journal = {Photosynth Res},
volume = {103},
number = {3},
pages = {183-94},
abstract = {CP12, a small intrinsically unstructured protein, plays an important role in the regulation of the Calvin cycle by forming a complex with phosphoribulokinase (PRK) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). An extensive search in databases revealed 129 protein sequences from, higher plants, mosses and liverworts, different groups of eukaryotic algae and cyanobacteria. CP12 was identified throughout the Plantae, apart from in the Prasinophyceae. Within the Chromalveolata, two putative CP12 proteins have been found in the genomes of the diatom Thalassiosira pseudonana and the haptophyte Emiliania huxleyi, but specific searches in further chromalveolate genomes or EST datasets did not reveal any CP12 sequences in other Prymnesiophyceae, Dinophyceae or Pelagophyceae. A species from the Euglenophyceae within the Excavata also appeared to lack CP12. Phylogenetic analysis showed a clear separation into a number of higher taxonomic clades and among different forms of CP12 in higher plants. Cyanobacteria, Chlorophyceae, Rhodophyta and Glaucophyceae, Bryophyta, and the CP12-3 forms in higher plants all form separate clades. The degree of disorder of CP12 was higher in higher plants than in the eukaryotic algae and cyanobacteria apart from the green algal class Mesostigmatophyceae, which is ancestral to the streptophytes. This suggests that CP12 has evolved to become more flexible and possibly take on more general roles. Different features of the CP12 sequences in the different taxonomic groups and their potential functions and interactions in the Calvin cycle are discussed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2009
Tyagi, Manoj; Bornot, Aurélie; Offmann, Bernard; de Brevern, Alexandre G
Analysis of loop boundaries using different local structure assignment methods Article de journal
Dans: Protein Sci, vol. 18, no. 9, p. 1869-81, 2009.
@article{Tyagi:2009ab,
title = {Analysis of loop boundaries using different local structure assignment methods},
author = {Manoj Tyagi and Aurélie Bornot and Bernard Offmann and Alexandre G {de Brevern}},
doi = {10.1002/pro.198},
year = {2009},
date = {2009-09-01},
journal = {Protein Sci},
volume = {18},
number = {9},
pages = {1869-81},
abstract = {Loops connect regular secondary structures. In many instances, they are known to play important biological roles. Analysis and prediction of loop conformations depend directly on the definition of repetitive structures. Nonetheless, the secondary structure assignment methods (SSAMs) often lead to divergent assignments. In this study, we analyzed, both structure and sequence point of views, how the divergence between different SSAMs affect boundary definitions of loops connecting regular secondary structures. The analysis of SSAMs underlines that no clear consensus between the different SSAMs can be easily found. Because these latter greatly influence the loop boundary definitions, important variations are indeed observed, that is, capping positions are shifted between different SSAMs. On the other hand, our results show that the sequence information in these capping regions are more stable than expected, and, classical and equivalent sequence patterns were found for most of the SSAMs. This is, to our knowledge, the most exhaustive survey in this field as (i) various databank have been used leading to similar results without implication of protein redundancy and (ii) the first time various SSAMs have been used. This work hence gives new insights into the difficult question of assignment of repetitive structures and addresses the issue of loop boundaries definition. Although SSAMs give very different local structure assignments capping sequence patterns remain efficiently stable.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Tyagi, Manoj; Bornot, Aurélie; Offmann, Bernard; de Brevern, Alexandre G
Protein short loop prediction in terms of a structural alphabet Article de journal
Dans: Comput Biol Chem, vol. 33, no. 4, p. 329-33, 2009.
@article{Tyagi:2009aa,
title = {Protein short loop prediction in terms of a structural alphabet},
author = {Manoj Tyagi and Aurélie Bornot and Bernard Offmann and Alexandre G {de Brevern}},
doi = {10.1016/j.compbiolchem.2009.06.002},
year = {2009},
date = {2009-08-01},
journal = {Comput Biol Chem},
volume = {33},
number = {4},
pages = {329-33},
abstract = {Loops connect regular secondary structures. In many instances, they are known to play crucial biological roles. To bypass the limitation of secondary structure description, we previously defined a structural alphabet composed of 16 structural prototypes, called Protein Blocks (PBs). It leads to an accurate description of every region of 3D protein backbones and has been used in local structure prediction. In the present study, we used our structural alphabet to predict the loops connecting two repetitive structures. Thus, we showed interest to take into account the flanking regions, leading to prediction rate improvement up to 19.8%, but we also underline the sensitivity of such an approach. This research can be used to propose different structures for the loops and to probe and sample their flexibility. It is a useful tool for ab initio loop prediction and leads to insights into flexible docking approach.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sandhya, Sankaran; Rani, Saane Sudha; Pankaj, Barah; Govind, Madabosse Kande; Offmann, Bernard; Srinivasan, Narayanaswamy; Sowdhamini, Ramanathan
Length variations amongst protein domain superfamilies and consequences on structure and function Article de journal
Dans: PLoS One, vol. 4, no. 3, p. e4981, 2009.
@article{sandhya2009length,
title = {Length variations amongst protein domain superfamilies and consequences on structure and function},
author = {Sankaran Sandhya and Saane Sudha Rani and Barah Pankaj and Madabosse Kande Govind and Bernard Offmann and Narayanaswamy Srinivasan and Ramanathan Sowdhamini},
url = {https://doi.org/10.1371/journal.pone.0004981},
doi = {10.1371/journal.pone.0004981},
year = {2009},
date = {2009-01-01},
journal = {PLoS One},
volume = {4},
number = {3},
pages = {e4981},
publisher = {Public Library of Science},
abstract = {Background: Related protein domains of a superfamily can be specified by proteins of diverse lengths. The structural and functional implications of indels in a domain scaffold have been examined. Methodology: In this study, domain superfamilies with large length variations (more than 30% difference from average domain size, referred as 'length-deviant' superfamilies and 'length-rigid' domain superfamilies (<10% length difference from average domain size) were analyzed for the functional impact of such structural differences. Our delineated dataset, derived from an objective algorithm, enables us to address indel roles in the presence of peculiar structural repeats, functional variation, protein-protein interactions and to examine 'domain contexts' of proteins tolerant to large length variations. Amongst the top-10 length-deviant superfamilies analyzed, we found that 80% of length-deviant superfamilies possess distant internal structural repeats and nearly half of them acquired diverse biological functions. In general, length-deviant superfamilies have higher chance, than length-rigid superfamilies, to be engaged in internal structural repeats. We also found that approximately 40% of length-deviant domains exist as multi-domain proteins involving interactions with domains from the same or other superfamilies. Indels, in diverse domain superfamilies, were found to participate in the accretion of structural and functional features amongst related domains. With specific examples, we discuss how indels are involved directly or indirectly in the generation of oligomerization interfaces, introduction of substrate specificity, regulation of protein function and stability. Conclusions: Our data suggests a multitude of roles for indels that are specialized for domain members of different domain superfamilies. These specialist roles that we observe and trends in the extent of length variation could influence decision making in modeling of new superfamily members. Likewise, the observed limits of length variation, specific for each domain superfamily would be particularly relevant in the choice of alignment length search filters commonly applied in protein sequence analysis.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2008
Thangudu, Ratna R; Manoharan, Malini; Srinivasan, N; Cadet, Frédéric; Sowdhamini, R; Offmann, Bernard
Analysis on conservation of disulphide bonds and their structural features in homologous protein domain families Article de journal
Dans: BMC Struct Biol, vol. 8, p. 55, 2008.
@article{Thangudu:2008aa,
title = {Analysis on conservation of disulphide bonds and their structural features in homologous protein domain families},
author = {Ratna R Thangudu and Malini Manoharan and N Srinivasan and Frédéric Cadet and R Sowdhamini and Bernard Offmann},
doi = {10.1186/1472-6807-8-55},
year = {2008},
date = {2008-12-01},
journal = {BMC Struct Biol},
volume = {8},
pages = {55},
abstract = {BACKGROUND: Disulphide bridges are well known to play key roles in stability, folding and functions of proteins. Introduction or deletion of disulphides by site-directed mutagenesis have produced varying effects on stability and folding depending upon the protein and location of disulphide in the 3-D structure. Given the lack of complete understanding it is worthwhile to learn from an analysis of extent of conservation of disulphides in homologous proteins. We have also addressed the question of what structural interactions replaces a disulphide in a homologue in another homologue.
RESULTS: Using a dataset involving 34,752 pairwise comparisons of homologous protein domains corresponding to 300 protein domain families of known 3-D structures, we provide a comprehensive analysis of extent of conservation of disulphide bridges and their structural features. We report that only 54% of all the disulphide bonds compared between the homologous pairs are conserved, even if, a small fraction of the non-conserved disulphides do include cytoplasmic proteins. Also, only about one fourth of the distinct disulphides are conserved in all the members in protein families. We note that while conservation of disulphide is common in many families, disulphide bond mutations are quite prevalent. Interestingly, we note that there is no clear relationship between sequence identity between two homologous proteins and disulphide bond conservation. Our analysis on structural features at the sites where cysteines forming disulphide in one homologue are replaced by non-Cys residues show that the elimination of a disulphide in a homologue need not always result in stabilizing interactions between equivalent residues.
CONCLUSION: We observe that in the homologous proteins, disulphide bonds are conserved only to a modest extent. Very interestingly, we note that extent of conservation of disulphide in homologous proteins is unrelated to the overall sequence identity between homologues. The non-conserved disulphides are often associated with variable structural features that were recruited to be associated with differentiation or specialisation of protein function.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
RESULTS: Using a dataset involving 34,752 pairwise comparisons of homologous protein domains corresponding to 300 protein domain families of known 3-D structures, we provide a comprehensive analysis of extent of conservation of disulphide bridges and their structural features. We report that only 54% of all the disulphide bonds compared between the homologous pairs are conserved, even if, a small fraction of the non-conserved disulphides do include cytoplasmic proteins. Also, only about one fourth of the distinct disulphides are conserved in all the members in protein families. We note that while conservation of disulphide is common in many families, disulphide bond mutations are quite prevalent. Interestingly, we note that there is no clear relationship between sequence identity between two homologous proteins and disulphide bond conservation. Our analysis on structural features at the sites where cysteines forming disulphide in one homologue are replaced by non-Cys residues show that the elimination of a disulphide in a homologue need not always result in stabilizing interactions between equivalent residues.
CONCLUSION: We observe that in the homologous proteins, disulphide bonds are conserved only to a modest extent. Very interestingly, we note that extent of conservation of disulphide in homologous proteins is unrelated to the overall sequence identity between homologues. The non-conserved disulphides are often associated with variable structural features that were recruited to be associated with differentiation or specialisation of protein function.
Reddy, Chilamakuri C S; Shameer, Khader; Offmann, Bernard; Sowdhamini, Ramanathan
PURE: a webserver for the prediction of domains in unassigned regions in proteins Article de journal
Dans: BMC Bioinformatics, vol. 9, p. 281, 2008.
@article{Reddy:2008aa,
title = {PURE: a webserver for the prediction of domains in unassigned regions in proteins},
author = {Chilamakuri C S Reddy and Khader Shameer and Bernard Offmann and Ramanathan Sowdhamini},
doi = {10.1186/1471-2105-9-281},
year = {2008},
date = {2008-06-01},
journal = {BMC Bioinformatics},
volume = {9},
pages = {281},
abstract = {BACKGROUND: Protein domains are the structural and functional units of proteins. The ability to parse proteins into different domains is important for effective classification, understanding of protein structure, function, and evolution and is hence biologically relevant. Several computational methods are available to identify domains in the sequence. Domain finding algorithms often employ stringent thresholds to recognize sequence domains. Identification of additional domains can be tedious involving intense computation and manual intervention but can lead to better understanding of overall biological function. In this context, the problem of identifying new domains in the unassigned regions of a protein sequence assumes a crucial importance. RESULTS: We had earlier demonstrated that accumulation of domain information of sequence homologues can substantially aid prediction of new domains. In this paper, we propose a computationally intensive, multi-step bioinformatics protocol as a web server named as PURE (Prediction of Unassigned REgions in proteins) for the detailed examination of stretches of unassigned regions in proteins. Query sequence is processed using different automated filtering steps based on length, presence of coiled-coil regions, transmembrane regions, homologous sequences and percentage of secondary structure content. Later, the filtered sequence segments and their sequence homologues are fed to PSI-BLAST, cd-hit and Hmmpfam. Data from the various programs are integrated and information regarding the probable domains predicted from the sequence is reported. CONCLUSION: We have implemented PURE protocol as a web server for rapid and comprehensive analysis of unassigned regions in the proteins. This server integrates data from different programs and provides information about the domains encoded in the unassigned regions.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Sandhya, Sankaran; Pankaj, Barah; Govind, Madabosse Kande; Offmann, Bernard; Srinivasan, Narayanaswamy; Sowdhamini, Ramanathan
CUSP: an algorithm to distinguish structurally conserved and unconserved regions in protein domain alignments and its application in the study of large length variations Article de journal
Dans: BMC Struct Biol, vol. 8, p. 28, 2008.
@article{Sandhya:2008aa,
title = {CUSP: an algorithm to distinguish structurally conserved and unconserved regions in protein domain alignments and its application in the study of large length variations},
author = {Sankaran Sandhya and Barah Pankaj and Madabosse Kande Govind and Bernard Offmann and Narayanaswamy Srinivasan and Ramanathan Sowdhamini},
doi = {10.1186/1472-6807-8-28},
year = {2008},
date = {2008-05-01},
journal = {BMC Struct Biol},
volume = {8},
pages = {28},
abstract = {BACKGROUND: Distantly related proteins adopt and retain similar structural scaffolds despite length variations that could be as much as two-fold in some protein superfamilies. In this paper, we describe an analysis of indel regions that accommodate length variations amongst related proteins. We have developed an algorithm CUSP, to examine multi-membered PASS2 superfamily alignments to identify indel regions in an automated manner. Further, we have used the method to characterize the length, structural type and biochemical features of indels in related protein domains. RESULTS: CUSP, examines protein domain structural alignments to distinguish regions of conserved structure common to related proteins from structurally unconserved regions that vary in length and type of structure. On a non-redundant dataset of 353 domain superfamily alignments from PASS2, we find that 'length- deviant' protein superfamilies show > 30% length variation from their average domain length. 60% of additional lengths that occur in indels are short-length structures (< 5 residues) while 6% of indels are > 15 residues in length. Structural types in indels also show class-specific trends. CONCLUSION: The extent of length variation varies across different superfamilies and indels show class-specific trends for preferred lengths and structural types. Such indels of different lengths even within a single protein domain superfamily could have structural and functional consequences that drive their selection, underlying their importance in similarity detection and computational modelling. The availability of systematic algorithms, like CUSP, should enable decision making in a domain superfamily-specific manner.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Tyagi, M; de Brevern, A G; Srinivasan, N; Offmann, Bernard
Protein structure mining using a structural alphabet Article de journal
Dans: Proteins, vol. 71, no. 2, p. 920-37, 2008.
@article{Tyagi:2008aa,
title = {Protein structure mining using a structural alphabet},
author = {M Tyagi and A G {de Brevern} and N Srinivasan and Bernard Offmann},
doi = {10.1002/prot.21776},
year = {2008},
date = {2008-05-01},
journal = {Proteins},
volume = {71},
number = {2},
pages = {920-37},
abstract = {We present a comprehensive evaluation of a new structure mining method called PB-ALIGN. It is based on the encoding of protein structure as 1D sequence of a combination of 16 short structural motifs or protein blocks (PBs). PBs are short motifs capable of representing most of the local structural features of a protein backbone. Using derived PB substitution matrix and simple dynamic programming algorithm, PB sequences are aligned the same way amino acid sequences to yield structure alignment. PBs are short motifs capable of representing most of the local structural features of a protein backbone. Alignment of these local features as sequence of symbols enables fast detection of structural similarities between two proteins. Ability of the method to characterize and align regions beyond regular secondary structures, for example, N and C caps of helix and loops connecting regular structures, puts it a step ahead of existing methods, which strongly rely on secondary structure elements. PB-ALIGN achieved efficiency of 85% in extracting true fold from a large database of 7259 SCOP domains and was successful in 82% cases to identify true super-family members. On comparison to 13 existing structure comparison/mining methods, PB-ALIGN emerged as the best on general ability test dataset and was at par with methods like YAKUSA and CE on nontrivial test dataset. Furthermore, the proposed method performed well when compared to flexible structure alignment method like FATCAT and outperforms in processing speed (less than 45 s per database scan). This work also establishes a reliable cut-off value for the demarcation of similar folds. It finally shows that global alignment scores of unrelated structures using PBs follow an extreme value distribution. PB-ALIGN is freely available on web server called Protein Block Expert (PBE) at http://bioinformatics.univ-reunion.fr/PBE/.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2007
Chilamakuri, Sekhar Reddy C; Khader, Shameer; Offmann, Bernard; Sowdhamini, Ramanathan
PURE: a web server for querying the relationship between pre-existing domains and unassigned regions in proteins Article de journal
Dans: Nature Protocols, 2007.
@article{chilamakuri2007pure,
title = {PURE: a web server for querying the relationship between pre-existing domains and unassigned regions in proteins},
author = {Sekhar Reddy C Chilamakuri and Shameer Khader and Bernard Offmann and Ramanathan Sowdhamini},
doi = {doi:10.1038/nprot.2007.486},
year = {2007},
date = {2007-11-01},
journal = {Nature Protocols},
publisher = {Nature Publishing Group},
abstract = {Protein domains are the structural and functional units of proteins. The ability to parse proteins into different domains is important for effective classification, understanding of protein structure, function and evolution and is hence biologically relevant. Several computational methods are available to identify domains in the sequence. Domain finding algorithms often employ stringent thresholds to recognize sequence domains. Identification of additional domains can be tedious involving intense computation and manual intervention but can lead to better understanding of overall biological function. In this context, the problem of identifying new domains in the unassigned regions of a protein sequence assumes a crucial importance. We report the availability of a convenient server for the domain prediction in unassigned regions in proteins (PURE) which can be accessed at http://caps.ncbs.res.in/PURE/ year = 2007},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Thangudu, Ratna R; Sharma, Priyanka; Srinivasan, N; Offmann, Bernard
Analycys: a database for conservation and conformation of disulphide bonds in homologous protein domains Article de journal
Dans: Proteins, vol. 67, no. 2, p. 255-61, 2007.
@article{Thangudu:2007aa,
title = {Analycys: a database for conservation and conformation of disulphide bonds in homologous protein domains},
author = {Ratna R Thangudu and Priyanka Sharma and N Srinivasan and Bernard Offmann},
doi = {10.1002/prot.21318},
year = {2007},
date = {2007-05-01},
journal = {Proteins},
volume = {67},
number = {2},
pages = {255-61},
abstract = {Disulphide bonds in proteins are known to play diverse roles ranging from folding to structure to function. Thorough knowledge of the conservation status and structural state of the disulphide bonds will help in understanding of the differences in homologous proteins. Here we present a database for the analysis of conservation and conformation of disulphide bonds in SCOP structural families. This database has a wide range of applications including mapping of disulphide bond mutation patterns, identification of disulphide bonds important for folding and stabilization, modeling of protein tertiary structures and in protein engineering. The database can be accessed at: http://bioinformatics.univ-reunion.fr/analycys/.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Bornot, Aurélie; Offmann, Bernard; de Brevern, Alexandre
How flexible protein structures are? New questions on the protein structure plasticity. Article de journal
Dans: Bioforum Europe, no. 11, p. 24, 2007.
@article{bornot2007flexible,
title = {How flexible protein structures are? New questions on the protein structure plasticity.},
author = {Aurélie Bornot and Bernard Offmann and Alexandre {de Brevern}},
year = {2007},
date = {2007-01-01},
journal = {Bioforum Europe},
number = {11},
pages = {24},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Swapna, LS; Offmann, Bernard; Srinivasan, N
Evolutionary dynamics of protein-protein interactions. A case study using the DJ-1/PfpI family of enzyme. Ouvrage
John Wiley & Sons, Inc., 2007.
@book{swapna2007evolutionary,
title = {Evolutionary dynamics of protein-protein interactions. A case study using the DJ-1/PfpI family of enzyme.},
author = {LS Swapna and Bernard Offmann and N Srinivasan},
year = {2007},
date = {2007-01-01},
journal = {Knowledge Discovery in Bioinformatics: Techniques, Methods, and Applications},
pages = {209--231},
publisher = {John Wiley & Sons, Inc.},
keywords = {},
pubstate = {published},
tppubtype = {book}
}
Offmann, Bernard; Tyagi, Manoj; de Brevern, Alexandre G.
Local Protein Structures Article de journal
Dans: Current Bioinformatics, vol. 2, no. 3, p. 165-202, 2007, ISSN: 1574-8936/2212-392X.
@article{BernardOffmann2007,
title = {Local Protein Structures},
author = {Bernard Offmann and Manoj Tyagi and Alexandre G. {de Brevern}},
url = {http://www.eurekaselect.com/node/78709/article},
doi = {10.2174/157489307781662105},
issn = {1574-8936/2212-392X},
year = {2007},
date = {2007-01-01},
journal = {Current Bioinformatics},
volume = {2},
number = {3},
pages = {165-202},
abstract = {Protein structures are classically described as composed of two regular states, the α-helices and the β-strands and one non-regular and variable state, the coil. Nonetheless, this simple definition of secondary structures hides numerous limitations. In fact, the rules for secondary structure assignment are complex. Thus, numerous assignment methods based on different criteria have emerged leading to heterogeneous and diverging results. In the same way, 3 states may over-simplify the description of protein structure; 50% of all residues, i.e., the coil, are not genuinely described even when it encompass precise local protein structures. Description of local protein structures have hence focused on the elaboration of complete sets of small prototypes or structural alphabets, able to analyze local protein structures and to approximate every part of the protein backbone. They have also been used to predict the protein backbone conformation and in ab initio/ de novo methods. In this paper, we review different approaches towards the description of local structures, mainly through their description in terms of secondary structures and in terms of structural alphabets. We provide some insights into their potential applications.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2006
Gardebien, Fabrice; Thangudu, Rajesh R; Gontero, Brigitte; Offmann, Bernard
Construction of a 3D model of CP12, a protein linker Article de journal
Dans: J Mol Graph Model, vol. 25, no. 2, p. 186-95, 2006.
@article{Gardebien:2006aa,
title = {Construction of a 3D model of CP12, a protein linker},
author = {Fabrice Gardebien and Rajesh R Thangudu and Brigitte Gontero and Bernard Offmann},
doi = {10.1016/j.jmgm.2005.12.003},
year = {2006},
date = {2006-10-01},
journal = {J Mol Graph Model},
volume = {25},
number = {2},
pages = {186-95},
abstract = {The chloroplast protein CP12 is known to play a leading role in a complex formation with the enzymes GAPDH and PRK. As a preliminary step towards the understanding of the complex formation mechanism and the exact role of this protein linker, a comparative modelling of the CP12 protein of the green alga Chlamydomonas reinhardtii was performed. Because of the very few structural information and poor template similarities, the derivation of the model consisted in an iterative trial-and-error procedure using the comparative modelling program MODELLER, the following three structure validation programs PROCHECK, PROSA, and WHATIF, and molecular mechanics energy refinement of the model using the program CHARMM. The analysis of the final model reveals a scaffold of key residues that is believed to be essential in the folding mechanism and that coincides with the residues conserved throughout the CP12 family. Our results suggest that this protein is a typical disordered protein. Finally, the various mechanisms by which the CP12 protein can self-interact or binds to other enzymes are discussed in light of its modelled structure and characteristics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Tyagi, Manoj; Gowri, Venkataraman S; Srinivasan, Narayanaswamy; de Brevern, Alexandre G; Offmann, Bernard
A substitution matrix for structural alphabet based on structural alignment of homologous proteins and its applications Article de journal
Dans: Proteins, vol. 65, no. 1, p. 32-9, 2006.
@article{Tyagi:2006aa,
title = {A substitution matrix for structural alphabet based on structural alignment of homologous proteins and its applications},
author = {Manoj Tyagi and Venkataraman S Gowri and Narayanaswamy Srinivasan and Alexandre G {de Brevern} and Bernard Offmann},
doi = {10.1002/prot.21087},
year = {2006},
date = {2006-10-01},
journal = {Proteins},
volume = {65},
number = {1},
pages = {32-9},
abstract = {Analysis of protein structures based on backbone structural patterns known as structural alphabets have been shown to be very useful. Among them, a set of 16 pentapeptide structural motifs known as protein blocks (PBs) has been identified and upon which backbone model of most protein structures can be built. PBs allows simplification of 3D space onto 1D space in the form of sequence of PBs. Here, for the first time, substitution probabilities of PBs in a large number of aligned homologous protein structures have been studied and are expressed as a simplified 16 x 16 substitution matrix. The matrix was validated by benchmarking how well it can align sequences of PBs rather like amino acid alignment to identify structurally equivalent regions in closely or distantly related proteins using dynamic programming approach. The alignment results obtained are very comparable to well established structure comparison methods like DALI and STAMP. Other interesting applications of the matrix have been investigated. We first show that, in variable regions between two superimposed homologous proteins, one can distinguish between local conformational differences and rigid-body displacement of a conserved motif by comparing the PBs and their substitution scores. Second, we demonstrate, with the example of aspartic proteinases, that PBs can be efficiently used to detect the lobe/domain flexibility in the multidomain proteins. Lastly, using protein kinase as an example, we identify regions of conformational variations and rigid body movements in the enzyme as it is changed to the active state from an inactive state.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Tyagi, M; Sharma, P; Swamy, C S; Cadet, F; Srinivasan, N; de Brevern, A G; Offmann, Bernard
Protein Block Expert (PBE): a web-based protein structure analysis server using a structural alphabet Article de journal
Dans: Nucleic Acids Res, vol. 34, no. Web Server issue, p. W119-23, 2006.
@article{Tyagi:2006ab,
title = {Protein Block Expert (PBE): a web-based protein structure analysis server using a structural alphabet},
author = {M Tyagi and P Sharma and C S Swamy and F Cadet and N Srinivasan and A G {de Brevern} and Bernard Offmann},
doi = {10.1093/nar/gkl199},
year = {2006},
date = {2006-07-01},
journal = {Nucleic Acids Res},
volume = {34},
number = {Web Server issue},
pages = {W119-23},
abstract = {Encoding protein 3D structures into 1D string using short structural prototypes or structural alphabets opens a new front for structure comparison and analysis. Using the well-documented 16 motifs of Protein Blocks (PBs) as structural alphabet, we have developed a methodology to compare protein structures that are encoded as sequences of PBs by aligning them using dynamic programming which uses a substitution matrix for PBs. This methodology is implemented in the applications available in Protein Block Expert (PBE) server. PBE addresses common issues in the field of protein structure analysis such as comparison of proteins structures and identification of protein structures in structural databanks that resemble a given structure. PBE-T provides facility to transform any PDB file into sequences of PBs. PBE-ALIGNc performs comparison of two protein structures based on the alignment of their corresponding PB sequences. PBE-ALIGNm is a facility for mining SCOP database for similar structures based on the alignment of PBs. Besides, PBE provides an interface to a database (PBE-SAdb) of preprocessed PB sequences from SCOP culled at 95% and of all-against-all pairwise PB alignments at family and superfamily levels. PBE server is freely available at http://bioinformatics.univ-reunion.fr/PBE/.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2005
Thangudu, Ratna Rajesh; Vinayagam, A; Pugalenthi, G; Manonmani, A; Offmann, Bernard; Sowdhamini, R
Native and modeled disulfide bonds in proteins: knowledge-based approaches toward structure prediction of disulfide-rich polypeptides Article de journal
Dans: Proteins, vol. 58, no. 4, p. 866-79, 2005.
@article{Thangudu:2005aa,
title = {Native and modeled disulfide bonds in proteins: knowledge-based approaches toward structure prediction of disulfide-rich polypeptides},
author = {Ratna Rajesh Thangudu and A Vinayagam and G Pugalenthi and A Manonmani and Bernard Offmann and R Sowdhamini},
doi = {10.1002/prot.20369},
year = {2005},
date = {2005-03-01},
journal = {Proteins},
volume = {58},
number = {4},
pages = {866-79},
abstract = {Structure prediction and three-dimensional modeling of disulfide-rich systems are challenging due to the limited number of such folds in the structural databank. We exploit the stereochemical compatibility of substructures in known protein structures to accommodate disulfide bonds in predicting the structures of disulfide-rich polypeptides directly from disulfide connectivity pattern and amino acid sequence in the absence of structural homologs and any other structural information. This knowledge-based approach is illustrated using structure prediction of 40 nonredundant bioactive disulfide-rich polypeptides such as toxins, growth factors, and endothelins available in the structural databank. The polypeptide conformation could be predicted in 35 out of 40 nonredundant entries (87%). Nonhomologous templates could be identified and models could be obtained within 2 A deviation from the query in 29 peptides (72%). This procedure can be accessed from the World Wide Web (http://www.ncbs.res.in/ approximately faculty/mini/dsdbase/dsdbase.html).},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2003
Besnard, G; Pinçon, G; D'Hont, A; Hoarau, J-Y; Cadet, Frédéric; Offmann, Bernard
Characterisation of the phosphoenolpyruvate carboxylase gene family in sugarcane (Saccharum spp.) Article de journal
Dans: Theoretical and Applied Genetics, vol. 107, no. 3, p. 470-8, 2003.
@article{Besnard:2003aa,
title = {Characterisation of the phosphoenolpyruvate carboxylase gene family in sugarcane (Saccharum spp.)},
author = {G Besnard and G Pinçon and A D'Hont and J-Y Hoarau and Frédéric Cadet and Bernard Offmann},
doi = {10.1007/s00122-003-1268-2},
year = {2003},
date = {2003-08-01},
journal = {Theoretical and Applied Genetics},
volume = {107},
number = {3},
pages = {470-8},
abstract = {Phosphoenolpyruvate carboxylases (PEPCs) are encoded by a small multigenic family. In order to characterise this gene family in sugarcane, seven DNA fragments displaying a high homology with grass PEPC genes were isolated using polymerase chain reaction-based cloning. A phylogenetic study revealed the existence of four main PEPC gene lineages in grasses and particularly in sugarcane. Moreover, this analysis suggests that grass C4 PEPC has likely derived from a root pre-existing isoform in an ancestral species. Using the Northern-dot-blot method, we studied the expression of the four PEPC gene classes in sugarcane cv. R570. We confirmed that transcript accumulation of the C4 PEPC gene (ppc-C4) mainly occurs in the green leaves and is light-induced. We also showed that another member of this gene family (ppc-aR) is more highly transcribed in the roots. The constitutive expression for a previously characterised gene (ppc-aL2) was confirmed. Lastly, the transcript accumulation of the fourth PEPC gene class (ppc-aL1) was not revealed. Length polymorphism in non-coding regions for three PEPC gene lineages enabled us to develop sequence-tagged site PEPC markers in sugarcane. We analysed the segregation of PEPC fragments in self-pollinated progenies of cv. R570 and found co-segregating fragments for two PEPC gene lineages. This supports the hypothesis that diversification of the PEPC genes involved duplications, probably in tandem.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Offmann, Bernard; Raboin, Louis Marie; Notaise, Julien
Segregation of resistance to Xanthomonas albilineans in a sugarcane progeny Proceedings Article
Dans: p. 113–124, Bourbon sciences, 2003.
@inproceedings{offmann2003segregation,
title = {Segregation of resistance to Xanthomonas albilineans in a sugarcane progeny},
author = {Bernard Offmann and Louis Marie Raboin and Julien Notaise},
year = {2003},
date = {2003-01-01},
journal = {The Journal of Nature},
volume = {15},
number = {1},
pages = {113--124},
publisher = {Bourbon sciences},
keywords = {},
pubstate = {published},
tppubtype = {inproceedings}
}
2002
Hoarau, J-Y; Grivet, L; Offmann, Bernard; Raboin, L-M; Diorflar, J-P; Payet, J; Hellmann, M; D'Hont, A; Glaszmann, J-C
Genetic dissection of a modern sugarcane cultivar ( Saccharum spp.).II. Detection of QTLs for yield components Article de journal
Dans: Theoretical and Applied Genetics, vol. 105, no. 6-7, p. 1027-1037, 2002.
@article{Hoarau:2002aa,
title = {Genetic dissection of a modern sugarcane cultivar ( Saccharum spp.).II. Detection of QTLs for yield components},
author = {J-Y Hoarau and L Grivet and Bernard Offmann and L-M Raboin and J-P Diorflar and J Payet and M Hellmann and A D'Hont and J-C Glaszmann},
doi = {10.1007/s00122-002-1047-5},
year = {2002},
date = {2002-11-01},
journal = {Theoretical and Applied Genetics},
volume = {105},
number = {6-7},
pages = {1027-1037},
abstract = {The genetics of current sugarcane cultivars ( Saccharum spp.) is outstandingly complex, due to a high ploidy level and an interspecific origin which leads to the presence of numerous chromosomes belonging to two ancestral genomes. In order to analyse the inheritance of quantitative traits, we have undertaken an extensive Quantitative Trait Allele (QTA) mapping study based on a population of 295 progenies derived from the selfing of cultivar R570, using about 1,000 AFLP markers scattered on about half of the genome. The population was evaluated in a replicated trial for four basic yield components, plant height, stalk number, stalk diameter and brix, in two successive crop-cycles. Forty putative QTAs were found for the four traits at P = 5 x 10(-3), of which five appeared in both years. Their individual size ranged between 3 and 7% of the whole variation. The stability across years was improved when limiting threshold stringency. All these results depict the presence in the genome of numerous QTAs, with little effects, fluctuating slightly across cycles, on the verge to being perceptible given the experimental resolution. Epistatic interactions were also explored and 41 independent di-genic interactions were found at P = (5 x 10(-3))(2). Altogether the putative genetic factors revealed here explain from 30 to 55% of the total phenotypic variance depending on the trait. The tentative assignment of some QTAs to the ancestral genomes showed a small majority of contributions as expected from the ancestral phenotypes. This is the first extensive QTL mapping study performed in cultivated sugarcane.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Besnard, Guillaume; Offmann, Bernard; Robert, Christine; Rouch, Claude; Cadet, Frédéric
Assessment of the C(4) phosphoenolpyruvate carboxylase gene diversity in grasses (Poaceae) Article de journal
Dans: Theoretical and Applied Genetics, vol. 105, no. 2-3, p. 404-412, 2002.
@article{Besnard:2002aa,
title = {Assessment of the C(4) phosphoenolpyruvate carboxylase gene diversity in grasses (Poaceae)},
author = {Guillaume Besnard and Bernard Offmann and Christine Robert and Claude Rouch and Frédéric Cadet},
doi = {10.1007/s00122-001-0851-7},
year = {2002},
date = {2002-08-01},
journal = {Theoretical and Applied Genetics},
volume = {105},
number = {2-3},
pages = {404-412},
abstract = {C(4) phosphoenolpyruvate carboxylase (PEPC) is a key enzyme in the C(4) photosynthetic pathway. To analyze the diversity of the corresponding gene in grasses, we designed PCR primers to specifically amplify C(4) PEPC cDNA fragments. Using RT-PCR, we generated partial PEPC cDNA sequences in several grasses displaying a C(4) photosynthetic pathway. All these sequences displayed a high homology (78-99%) with known grass C(4) PEPCs. PCR amplification did not occur in two grasses that display the C(3) photosynthetic pathway, and therefore we assumed that all generated sequences corresponded to C(4) PEPC transcripts. Based on one large cDNA segment, phylogenetic reconstruction enabled us to assess the relationships between 22 grass species belonging to the subfamilies Panicoideae, Arundinoideae and Chloridoideae. The phylogenetic relationships between species deduced from C(4) PEPC sequences were similar to those deduced from other molecular data. The sequence evolution of the C(4) PEPC isoform was faster than in the other PEPC isoforms. Finally, the utility of the C(4) PEPC gene phylogeny to study the evolution of C(4) photosynthesis in grasses is discussed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Offmann, Bernard; Cadet, Frédéric
Enzymes as structural tool in infrared spectroscopy Article de journal
Dans: Spectroscopy Letters, vol. 35, no. 4, p. 523-526, 2002.
@article{doi:10.1081/SL-120013888,
title = {Enzymes as structural tool in infrared spectroscopy},
author = {Bernard Offmann and Frédéric Cadet},
url = {https://www.tandfonline.com/doi/abs/10.1081/SL-120013888},
doi = {10.1081/SL-120013888},
year = {2002},
date = {2002-01-01},
journal = {Spectroscopy Letters},
volume = {35},
number = {4},
pages = {523-526},
publisher = {Taylor & Francis},
abstract = {The complexity of IR spectra of some compounds, particularly biological molecules, is a major obstacle in assigning or characterizing their IR bands. In this short communication, the potential use of enzymes for infrared bands characterization and assignments is demonstrated and discussed.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Offmann, Bernard; Cadet, Frédéric
Redox-active disulfides in a plant light switch: A Pbl problem* Article de journal
Dans: Biochemistry and Molecular Biology Education, vol. 30, no. 4, p. 249-254, 2002.
@article{https://doi.org/10.1002/bmb.2002.494030040069,
title = {Redox-active disulfides in a plant light switch: A Pbl problem*},
author = {Bernard Offmann and Frédéric Cadet},
url = {https://iubmb.onlinelibrary.wiley.com/doi/abs/10.1002/bmb.2002.494030040069},
doi = {https://doi.org/10.1002/bmb.2002.494030040069},
year = {2002},
date = {2002-01-01},
journal = {Biochemistry and Molecular Biology Education},
volume = {30},
number = {4},
pages = {249-254},
abstract = {A problem for students on the mode of activation of light-regulated NADP-dependent malate dehydrogenase is presented here as a series of questions requiring the interpretation of experimental data.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2001
Hoarau, J-Y; Offmann, Bernard; D'Hont, Angélique; Risterucci, A-M; Roques, D; Glaszmann, J-C; Grivet, Laurent
Genetic dissection of a modern sugarcane cultivar (Saccharum spp.). I. Genome mapping with AFLP markers Article de journal
Dans: Theoretical and Applied Genetics, vol. 103, no. 1, p. 84–97, 2001.
@article{hoarau2001genetic,
title = {Genetic dissection of a modern sugarcane cultivar (Saccharum spp.). I. Genome mapping with AFLP markers},
author = {J-Y Hoarau and Bernard Offmann and Angélique D'Hont and A-M Risterucci and D Roques and J-C Glaszmann and Laurent Grivet},
year = {2001},
date = {2001-01-01},
journal = {Theoretical and Applied Genetics},
volume = {103},
number = {1},
pages = {84--97},
publisher = {Springer-Verlag},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Raboin, Louis-Marie; Offmann, Bernard; Hoarau, Jean-Yves; Notaise, Julien; Costet, Laurent; Telismart, Hugues; Roques, Danièle; Rott, Philippe; Glaszmann, Jean-Christophe; D'Hont, Angélique
Undertaking genetic mapping of sugarcane smut resistance Proceedings Article
Dans: Proc S Afr Sug Technol Ass, p. 94–98, 2001.
@inproceedings{raboin2001undertaking,
title = {Undertaking genetic mapping of sugarcane smut resistance},
author = {Louis-Marie Raboin and Bernard Offmann and Jean-Yves Hoarau and Julien Notaise and Laurent Costet and Hugues Telismart and Danièle Roques and Philippe Rott and Jean-Christophe Glaszmann and Angélique D'Hont},
year = {2001},
date = {2001-01-01},
booktitle = {Proc S Afr Sug Technol Ass},
volume = {75},
pages = {94--98},
abstract = {Smut is one of the most important diseases of sugarcane and has a worldwide distribution. It can cause severe yield losses when a susceptible variety is grown in a smut infested area. Resistance is therefore a major concern for most sugarcane breeding centers. A study on the genetic determinism underlying sugarcane smut resistance was initiated. A genetic mapping strategy was cho- sen that focused on a cross between cultivar R 570 (resistant) and cultivar MQ 76/53 (highly susceptible) which showed a segregation for smut resistance in a preliminary field trial. An AFLP map is being constructed for both parents of the cross. At the same time, field trials and greenhouse experiments have begun using different artificial inoculation methods to assess the resistance of 200 individual progeny.This paper presents first results on smut occurrence among this progeny and correlations between segregating markers and re- sistance to smut. Other characters have also been observed (Brix, number of stalks, rust resistance). The possibility of iden- tifying the different components involved in smut resistance and the interest of locus specific markers (SSR, resistance gene analogs, etc) to refine the genetic map are discussed.},
keywords = {},
pubstate = {published},
tppubtype = {inproceedings}
}
Besnard, Guillaume; Offmann, Bernard; Robert, Christine; Baret, Pascal; Rouch, Claude; Cadet, Frédéric
Phosphoenolpyruvate carboxylase cDNA phylogeny to investigate the C4 photosynthetic pathway evolution in grasses Article de journal
Dans: Science Access, vol. 3, no. 1, 2001.
@article{besnard2001phosphoenolpyruvate,
title = {Phosphoenolpyruvate carboxylase cDNA phylogeny to investigate the C4 photosynthetic pathway evolution in grasses},
author = {Guillaume Besnard and Bernard Offmann and Christine Robert and Pascal Baret and Claude Rouch and Frédéric Cadet},
year = {2001},
date = {2001-01-01},
journal = {Science Access},
volume = {3},
number = {1},
publisher = {CSIRO},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2000
Offmann, Bernard
Caractérisation et analyse génétique de la résistance de la canne à sucre à Xanthomonas albilineans Thèse
La Réunion, 2000.
@phdthesis{offmann2000caracterisation,
title = {Caractérisation et analyse génétique de la résistance de la canne à sucre à Xanthomonas albilineans},
author = {Bernard Offmann},
year = {2000},
date = {2000-01-01},
school = {La Réunion},
keywords = {},
pubstate = {published},
tppubtype = {phdthesis}
}
1997
Offmann, Bernard
Cartographie génétique et recherche de QTLs chez un cultivar élite de canne à sucre (Saccharum spp) au moyen de marqueurs AFLP Thèse
Université de Paris VII, 1997.
@mastersthesis{offmann1997cartographie,
title = {Cartographie génétique et recherche de QTLs chez un cultivar élite de canne à sucre (Saccharum spp) au moyen de marqueurs AFLP},
author = {Bernard Offmann},
year = {1997},
date = {1997-07-01},
school = {Université de Paris VII},
keywords = {},
pubstate = {published},
tppubtype = {mastersthesis}
}
Cadet, Frédéric; Robert, Christine; Offmann, Bernard
Simultaneous Determination of Sugars by Multivariate Analysis Applied to Mid-Infrared Spectra of Biological Samples Article de journal
Dans: Appl. Spectrosc., vol. 51, no. 3, p. 369–375, 1997.
@article{Cadet:97,
title = {Simultaneous Determination of Sugars by Multivariate Analysis Applied to Mid-Infrared Spectra of Biological Samples},
author = {Frédéric Cadet and Christine Robert and Bernard Offmann},
url = {https://doi.org/10.1366/0003702971940224},
doi = {10.1366/0003702971940224},
year = {1997},
date = {1997-03-01},
urldate = {1997-03-01},
journal = {Appl. Spectrosc.},
volume = {51},
number = {3},
pages = {369--375},
publisher = {OSA},
abstract = {We have investigated the use of principal component analysis (PCA) to describe and assess mid-infrared spectral data obtained from complex biological samples containing sucrose, fructose, and glucose. The correlation coefficients between spectral data and chemical values of each variable (sucrose, glucose, fructose, total sugars, and reducing sugars) showed that in each case, axes 1, 3, 4, and 5 had the highest values. These values also indicated which axes each variable was mostly correlated with. The results also showed that the samples were distributed according to their sucrose concentrations (or total sugars) along a concentration gradient in the projection plan formed between axes 1 and 3. No clear discrimination according to concentration was observed with other factorial maps. Prediction equations that linked sucrose, fructose, glucose, total sugar, and reducing sugars concentrations to the spectral data were established by regression on the principal component. Very high correlation coefficients values between the first 10 axes and the chemical values were obtained (between 0.9757 and 0.998). From such aqueous biological samples containing a ternary mixture of sucrose, fructose, and glucose, it was possible to (1) identify the characteristic IR bands of these different sugars (and their combination: reducing sugars/total sugars) and (2) to specifically measure their concentrations with a relatively good accuracy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cadet, Frédéric; Offmann, Bernard
Direct Spectroscopic Sucrose Determination of Raw Sugar Cane Juices Article de journal
Dans: Journal of Agricultural and Food Chemistry, vol. 45, no. 1, p. 166-171, 1997.
@article{doi:10.1021/jf960700g,
title = {Direct Spectroscopic Sucrose Determination of Raw Sugar Cane Juices},
author = {Frédéric Cadet and Bernard Offmann},
url = {https://doi.org/10.1021/jf960700g},
doi = {10.1021/jf960700g},
year = {1997},
date = {1997-01-01},
journal = {Journal of Agricultural and Food Chemistry},
volume = {45},
number = {1},
pages = {166-171},
abstract = {A more accurate, less time-consuming, and nonpolluting spectroscopic method than the currently used HPLC or polarimetric methods is proposed for the routine quantitative determination of sucrose in complex biological samples. Opaque raw sugar cane juices representative of a sugar cane harvest are analyzed by Fourier transformed mid-infrared attenuated total reflectance, and the spectral data are processed by principal component analysis (PCA) and principal component regression (PCR). The most suitable region for the measurement of sucrose was found to be the 1250−800 cm-1 region. The spectroscopic representation of the first axis as assessed by PCA in this spectral region featured characteristic absorption bands of sucrose. By PCR on the spectral data from a calibration set, a prediction equation was established to predict sucrose content in unknown samples. Good overall predictions were obtained. The values of the predicted sucrose concentration were more accurate (bias = 0.041 g/100 mL) than those obtained by direct polarimetry (bias = −0.163 g/100 mL). The method is validated on a panel of 1267 samples representative of a sugar cane harvest.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
1996
Cadet, Frédéric; Offmann, Bernard
Evidence for Potassium-Sucrose Interaction in Biological Mid-Infrared Spectra by Multidimensional Analysis Article de journal
Dans: Spectroscopy letters, vol. 29, no. 7, p. 1353–1365, 1996.
@article{cadet1996evidence,
title = {Evidence for Potassium-Sucrose Interaction in Biological Mid-Infrared Spectra by Multidimensional Analysis},
author = {Frédéric Cadet and Bernard Offmann},
url = {https://doi.org/10.1080/00387019608007128},
doi = {10.1080/00387019608007128},
year = {1996},
date = {1996-01-01},
journal = {Spectroscopy letters},
volume = {29},
number = {7},
pages = {1353--1365},
publisher = {Taylor & Francis},
abstract = {Complex-formation between carbohydrates and cations could have important biological implications. In this work, Mid-Infrared spectra of pure sucrose solutions and of biological solutions containing sucrose and potassium ions (K+) were investigated by Principal Component Analysis (PGA). By direct examination of the Mid-Infrared spectra of the biological solutions containing K+ ions, no interactions between the cations and sucrose molecules could be observed. However, when the spectral pattern obtained by PCA and which is associated with sucrose, was examined, splitting and shifts in the characteristic absorption bands were observed owing to interactions between sucrose molecules and K+ ions. The 997 cm-1 peak which had a visible shoulder at 991 cm-1 and that is observed in pure solutions, was decomposed in the biological solutions into 3 distinct peaks at 1004, 996 and 990 cm-1. The two peaks centered at 1053 cm-1 were split into 3 peaks: 1060, 1051, 1045 cm-1. Hence by PCA, shoulders were characterized in biological solutions and more distinct peaks could be observed. These split and shift phenomena are similar to those obtained when crystalline sugar salts were investigated. This type of interaction, involving potassium ions and sucrose molecules, would be responsible for the storage of this cation which role is essential in plant metabolism.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cadet, Frédéric; Offmann, Bernard
Baseline correction applied to a biological: Mid-infrared spectra collection Article de journal
Dans: Spectroscopy letters, vol. 29, no. 4, p. 591–607, 1996.
@article{cadet1996baseline,
title = {Baseline correction applied to a biological: Mid-infrared spectra collection},
author = {Frédéric Cadet and Bernard Offmann},
url = {https://doi.org/10.1080/00387019608007054},
doi = {10.1080/00387019608007054},
year = {1996},
date = {1996-01-01},
journal = {Spectroscopy letters},
volume = {29},
number = {4},
pages = {591--607},
publisher = {Taylor & Francis Group},
abstract = {Near Infrared reflectance spectroscopy is now widely applied to predict the composition of various food products. On the other hand, in only few cases have Mid-infrared spectroscopy been reported to be used for the analysis of food products. However, this range is being more and more studied and important developments are being made. In infrared spectroscopy, random variations are often observed, adding an arbitrary constant to every absorbance value. With the aim of quantifying with the best precision obtainable, it is important that such spectra are corrected. The mean and the standard deviation of the absorbance at each wavelength has been calculated from a collection of biological spectra. The spectral regions where the standard deviation values were close to or equal to zero and which correspond to the most invariable zone have been chosen as reference. Uncertain variations from a sample spectrum are corrected by translation of the whole spectrum with respect to the mean reference spectrum. The 2092–2111.28 cm-1 zone has thus been chosen as reference zone. The corrected spectrum is obtained by: where Sc is the corrected spectrum, S the initial spectrum and cst is the difference in absorbance between the reference zone and the corresponding zone in the spectrum that is to be corrected. These constants vary between -1.14×10−3 and 5.35×10−3 Predictions have been established by Principal Component Regression on MIR spectra. It follows that the precision of the predicted values are sensibly improved. The bias values for the families of spectra that have not been corrected and that have been corrected are respectively -0.0637 and -0.0045 while the standard deviation values (SD) are 0.293 and 0.247 respectively. Such an automatic baseline correction method offers considerable advantages in cases where the precision of the quantitative measurements is primordial.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cadet, Frédéric; Offmann, Bernard
Gram-Schmidt Orthogonalization and Elimination of the Effect of Unwanted Component Spectra Applied to a Biological Mid-infrared Spectra Collection Article de journal
Dans: Spectroscopy letters, vol. 29, no. 5, p. 901–918, 1996.
@article{cadet1996gram,
title = {Gram-Schmidt Orthogonalization and Elimination of the Effect of Unwanted Component Spectra Applied to a Biological Mid-infrared Spectra Collection},
author = {Frédéric Cadet and Bernard Offmann},
url = {https://doi.org/10.1080/00387019608001620},
doi = {10.1080/00387019608001620},
year = {1996},
date = {1996-01-01},
journal = {Spectroscopy letters},
volume = {29},
number = {5},
pages = {901--918},
publisher = {Taylor & Francis},
abstract = {Near Infrared spectroscopy (NIR) is the most widely used technique for the analysis of major biochemical constituents in food products. The Mid-infrared range spectroscopy is also being more and more studied in the field of food analysis. This range was primarily applied to qualitative analysis of food components, but with the advent of new techniques such as Attenuated Total Reflectance (ATR) together with the possibility of combination with powerful micro-computers, MIR is now more and more used for quantitative analysis. In addition to baseline deformations, interference by unwanted compounds are major sources of problems that are encountered in analyses. We have previously proposed (Cadet et al., 1991) the use of multidimensional statistical analysis combined with Mid-FTIR spectroscopy for the prediction of sucrose concentrations in biological solutions containing three sugars: sucrose, fructose and glucose. In this paper, a least-squares method has been used to assess the elimination of the component spectra associated to the fructose (the unwanted components were first orthogonal zed and normalized by the Gram-Schmidt orthogonalization method). This procedure permitted the automatic subtraction of discriminant spectral patterns representative of fructose concentration before application of Principal Component Analysis (PCA) and Principal Component Regression (PCR). PCA was applied independently before and after spectral correction of the collections. It is found that when the factorial maps obtained before and after correction are compared, the elimination procedure improves significantly the classification of solutions according to their sucrose content. However the bias and standard deviation (SD) values that are associated with the sucrose content predicted values are not influenced by the correction method used: bias and SD are 3.62×10−2 and 3.097×10−1 before correction and after correction they were respectively 3.60×10−2 and 3.104×10−1. This could be explained by the fact that the presence of fructose in solution does not interfere with caracteristic absorption bands of sucrose and that sucrose and fructose concentrations are strongly correlated. The absorbtions bands of the spectral representation of the principal component, which is identified to be that associated with sucrose, are identical before and after correction.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Cadet, Frédéric; Offmann, Bernard
Extraction of Characteristic Bands of Sugars by Multidimensional Analysis of Their Infrared Spectra. Article de journal
Dans: Spectroscopy letters, vol. 29, no. 3, p. 523–536, 1996.
@article{cadet1996extraction,
title = {Extraction of Characteristic Bands of Sugars by Multidimensional Analysis of Their Infrared Spectra.},
author = {Frédéric Cadet and Bernard Offmann},
url = {https://doi.org/10.1080/00387019608006668},
doi = {10.1080/00387019608006668},
year = {1996},
date = {1996-01-01},
journal = {Spectroscopy letters},
volume = {29},
number = {3},
pages = {523--536},
publisher = {Taylor & Francis},
abstract = {Collected Mid-IR Attenuated Total Reflectance (ATR) spectra of various sugars were assessed by multidimensional statistical analysis. Through Principal Component Analysis (PCA) of collected spectra of various pure 10% sugar solutions and from the spectroscopic representation of the factorial axes, characteristic frequencies of monosaccharides and oligosaccharides were directly and automatically obtained within a few seconds. Monosugars are characterised by a hollow at 998 cm−1 and by a single unique major peak (1049 cm−1) in the 1075–1030 cm−1 region while oligosaccharides showed three characteristic bands in the same region, the major peak is shifted to 1033 cm−1. Owing to the glycosidic link vibrational motion, oligosaccharides are also characterised by a band at 998 cm−1. Spectroscopic representation of the axes issued of data from both sugar families (mixture of mono and oligosaccharides) is an average of the two individual spectral patterns. Predictive measures of concentrations of sugar solutions were performed by principal component regression (PCR) of the factorial coordinates and prediction equations were obtained. The predicted concentrations of standard 10% pure sugar solutions averaged 10. 069% and 9. 909% for monosugars and oligosugars respectively and a concentration of 10. 015% from the mixed set of data was obtained. The ability of these factorial coordinates to predict quantitative variable are good with correlation coefficients ranging from 97. 4% to 99. 9%.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Stagiaires encadrés :
- Thibaud LEPOIVRE, M1 Bioinformatique / Biostatistique, Nantes Université
- Ipek GUVENTÜRK, M2 Bioinformatique, Nantes Université
- Pauline DACLIN, M2 PAON, Nantes Université
- Rahamia SOILIHI MLANAOINDROU, Master de Bioinformatique, Nantes Université
- Julien LOUET, Master de Bioinformatique, Nantes Université
- Timothée SALZAT HERVOUETTE, Master de Bioinformatique, Nantes Université
- Damien RAT, Master de Bioinformatique, Nantes Université
- Anna CORNUAULT, Master de Bioinformatique, Nantes Université
- Justine PLOTEAU, Licence 3, Nantes Université